
Open Longevity. Как устроено старение и что с этим делать
Механическая связь с клеткой
Свойства матрикса сильно влияют на функционирование связанных с ним клеток, в частности на их развитие и дифференцировку. Клетки «чувствуют» степень жесткости матрикса при помощи различных механорецепторов: силы натяжения, сжатия и сдвига переводятся в биохимические сигналы. Этот процесс известен как механотрансдукция[11].
Растущая с возрастом жесткость матрикса и изменения в его структуре влияют на работу клеток, их способность к адгезии, снижение подвижности и другие аспекты их поведения41. Более того, повышение жесткости матрикса приводит к высвобождению клетками факторов роста, стимулирующих синтез компонентов матрикса и еще сильнее ее увеличивающих42 (рис. 3).
Многочисленные данные подтверждают, что клетки активно перестраивают окружающий их матрикс для поддержания оптимального уровня его жесткости43.
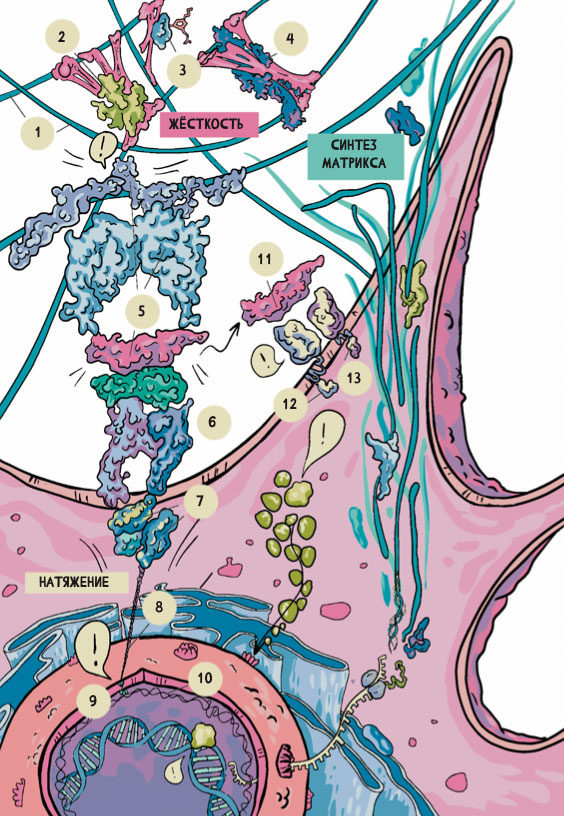
Рисунок 3. Механизмы повышения жесткости внеклеточного матрикса, силы механического натяжения и биохимические изменения, возникающие в ответ на возрастание жесткости матрикса
Поперечные сшивки матрикса, образуемые лизилоксидазой (LOX), тканевой трансглутаминазой (TG2), и конечные продукты гликирования – основные факторы, способствующие развитию патологической жесткости матрикса. Клетки детектируют повышение жесткости внеклеточного матрикса через фокальные контакты с матриксом, состоящие из интегринов и фокальных адгезионных комплексов. Сила натяжения актино-миозиновых волокон в клетках увеличивается в ответ на повышенную жесткость матрикса, усиленное натяжение волокон передается на матрикс, к которому они крепятся, и на ядра клеток. Опосредованное повышением жесткости матрикса увеличение силы натяжения актино-миозиновых волокон вызывает конформационные изменения в мультибелковом комплексе, связывающем трансформирующий фактор роста (TGF-β). Это приводит к высвобождению молекулы TGF-β, которая запускает синтез компонентов внеклеточного матрикса и повышение его жесткости.
Обозначения: 1 – внеклеточный матрикс; 2 – LOX; 3 – КПГ; 4 – TG2; 5 – латентный комплекс; 6 – интегрин; 7 – фокальная адгезия; 8 – актин; 9 – LINC; 10 – ламина; 11 – TGF-β; 12 – TbRII; 13 – TbRI42.
В 2010-х вышло несколько работ, показавших влияние жесткости матрикса на иммунную систему44. Есть основания полагать, что увеличение жесткости с возрастом ускоряет старение и гибель иммунных клеток, в частности тканевой популяции Т-лимфоцитов45. Судя по всему, одна из основных причин этого – механическое повреждение ядерной ламины при движении клетки сквозь матрикс46. Поврежденные же клетки становятся источником провоспалительных интерлейкинов, которые вызывают постоянное воспаление в тканях.
Подробнее о сшивках в матриксе
Изменения в структуре матрикса связывают в первую очередь с таким химическим процессом, как образование поперечных сшивок между его волокнами. Отметим несколько основных механизмов образования сшивок:
1. Ферментативное: с помощью трансглутаминазы47 и лизилоксидазы48 (рис. 2).
2. Неферментативное: 2а) гликирование белков; 2b) спонтанное расщепление по остатку аспарагина.
3. Карбамилирование.
Гликирование белков
Гликирование – неферментативное присоединение моносахаридов (глюкозы, фруктозы или их производных) к свободной аминогруппе белка с образованием химической сшивки (рис. 4). Химическое название данной реакции – реакция Майяра.
Если говорить простыми словами, это реакция «прилипания сахаров к белкам». Мы часто наблюдаем ее в быту: именно благодаря реакции Майяра при запекании получается аппетитная корочка на шашлыке или хлебе. Без дополнительного «поджаривания» реакция Майяра, впрочем, тоже протекает, просто медленнее – ровно это и происходит в нашем организме.
Вещества на корочке хлеба называются конечными продуктами гликирования (КПГ). Конечными в том смысле, что они получаются в конце реакции. И в том смысле, что назад дороги нет, корочка не превратится обратно в сырой хлеб.
Наличие КПГ в нашем теле – это на самом деле проблема, так как такие слипшиеся с моносахаридами белки перестают выполнять свои функции, а вернуться в нормальное функциональное состояние самостоятельно уже не могут. КПГ – это мусор, от которого необходимо избавиться, так как их накопление ведет к росту уровня воспаления в организме (к запуску экспрессии провоспалительных цитокинов)49.
В целом гликирование неполезно – это своего рода химическое «изнашивание» белков. Его даже можно считать маркером времени жизни белка50. Это случайный, стохастический процесс, происходящий в нашем теле безо всякой на то генетической программы.
Именно гликирование – виновник большинства повреждений тканей при сахарном диабете51. Кроме того, гликирование может приводить к нарушению функций митохондрий, их структуры и к окислительному стрессу52. Повышенное содержание КПГ обнаруживается в нервных клетках пациентов, страдающих болезнью Альцгеймера53, что указывает на участие гликирования в ее патогенезе41. Оно ускоряет процесс дегенерации нейронов, наблюдаемый при болезни Паркинсона54.
Гликирование же коллагена I типа делает его устойчивым к «перестройке» ферментами. Более того, между волокнами коллагена формируются «механические» сшивки, что увеличивает жесткость матрикса21, 55.
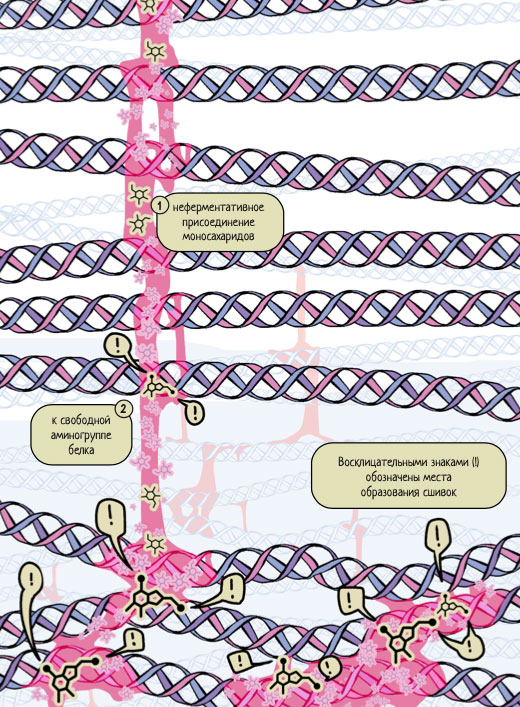
Рисунок 4. Схематичное изображение процесса гликирования белков: неферментативное присоединение моносахаридов (1) к свободной аминогруппе белка (2). Восклицательными знаками (!) обозначены места образования сшивок
Диагностика гликирования
Сейчас КПГ уже используются в клинической практике в качестве диагностического маркера для целого ряда заболеваний[12]56.
В частности, проводят анализ уровня гликирования белков в тканях, что важно для отслеживания изменений в долгосрочной перспективе. Это актуально и для коллагена, период полураспада которого – несколько лет, а уровень гликирования у здоровых людей в возрасте от 20 до 80 лет растет, хоть и незначительно, со временем61.
Обычно для кожи, как самой богатой коллагеном ткани, определяют уровень содержания глюкозепана (одного из КПГ). В ряде работ показана взаимосвязь роста количества глюкозепановых сшивок с сердечно-сосудистыми заболеваниями62, 63 и заболеваниями нервной системы62. Увеличение скорости гликирования коллагена говорит о развивающихся нарушениях, таких как кальцификация коронарных артерий, ретинопатия, нейропатия или нефропатия64.
В целом же наиболее перспективное направление развития предсказательной диагностики гликирования белков – комбинирование большого количества различных анализов в сочетании с приемами машинного обучения. Уже сейчас такие подходы65 показывают первые результаты в диагностике аутизма.
Еще один механизм образования сшивок
Он был открыт совсем недавно и включает в себя первоначальное спонтанное расщепление по остатку аспарагина в белке со ступенчатым расщеплением С-концевого сукцинимида, что в конечном счете приводит к образованию ангидрида. Нуклеофильная «атака» на этот ангидрид боковой аминогруппой лизина приводит к образованию ковалентной изопептидной связи.
Любой остаток аспарагина в белке, по-видимому, является потенциальным сайтом такого ковалентного сшивания66 (рис. 5).
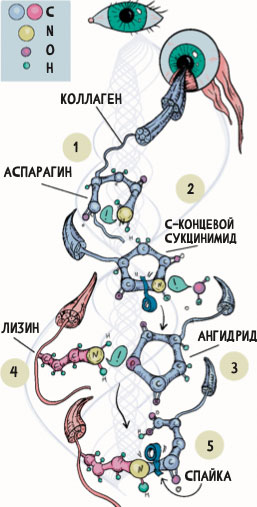
Рисунок 5. Один из механизмов образования сшивок в матриксе66
Первоначально остаток аспарагина (1) в белке подвергается спонтанному расщеплению, что приводит к формированию С-концевого сукцинимида (2), ступенчатое расщепление которого в конечном счете приводит к образованию ангидрида (3). Нуклеофильная «атака» на этот ангидрид боковой аминогруппой лизина (4) приводит к образованию ковалентной изопептидной связи (5). Любой остаток аспарагина в белке, по-видимому, – потенциальный сайт такого ковалентного сшивания.
Карбамилирование
Гликирование – не единственный тип химических реакций, ускоряющих старение матрикса. Группа французских исследователей показала67, что важным маркером старения является также карбамилирование – присоединение к аминогруппам белков остатков изоциановой кислоты, одного из продуктов распада мочевины (рис. 6).
При карбамилировании остатков лизина в долгоживущих белках матрикса накапливается гомоцитруллин. Его содержание напрямую коррелирует с продолжительностью жизни мышей, коров и людей.
Наличие гомоцитруллиновых остатков в молекулах коллагена приводит к дестабилизации его трехспиральной структуры и, как следствие, к снижению его устойчивости к температуре, действию металлопротеиназ и другим факторам деградации.
В итоге формально мы получаем тоже как бы сшивки. Но, в отличие от сшивок, возникших в результате процесса гликирования, они не делают волокна коллагена жестче. Напротив, коллаген становится более хрупким. Общее здесь одно – от такой сшивки организму тоже очень сложно избавиться самостоятельно.
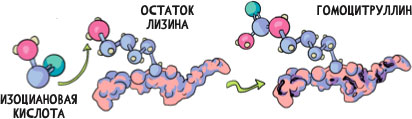
Рисунок 6. Химическая реакция карбамилирования лизина с образованием гомоцитруллина67
Ультрафиолет
Хроническое повреждение тканей происходит и при постоянном воздействии на них солнечного света. Основное действие ультрафиолета, по понятным причинам, приходится на ткани кожного покрова. В итоге кожа теряет упругость, появляются морщины.
Исследования показали68, что на молекулярном уровне фотостарение – это ровно то же самое старение, но в ускоренном виде. Воздействие ультрафиолета способствует появлению АФК в клетке. Это, в свою очередь, повышает уровень синтеза матриксных металлопротеиназ. А они уже фрагментируют коллагеновые волокна.
Фибробласты, закрепленные на волокнах коллагена, чувствуют снижение механического натяжения, а это – сигнал к дальнейшему синтезу металлопротеиназ и замедлению синтеза коллагена69. Механизм действия подобен порочному кругу: в результате старения ткани (в данном случае кожных покровов) в клетках повышается уровень АФК, что, в свою очередь, приводит к снижению механического натяжения – и снова повышает уровень АФК в клетках.
Также происходит замедление работы тирозиновых фосфатаз, в норме замедляющих развитие воспалительных реакций70.
В результате образуется положительная обратная связь, действие которой приводит к перманентному воспалению и ускоренному старению тканей кожного покрова. Опять «порочный круг».
Возрастное воспаление
О возрастном воспалении мы уже поговорили в первой главе. Это хроническое воспаление в пожилом возрасте. Оно усугубляет течение атеросклероза, диабета 2-го типа, болезни Альцгеймера и некоторых других заболеваний71.
КПГ активно привлекают макрофаги, которые принимают активное участие в воспалительных процессах. В результате концентрация макрофагов в ткани увеличивается и в ней развивается повышенный уровень воспаления72.
Ренин-ангиотензин-альдостероновая система
Известно, что с изменением структуры матрикса также связана активность РААС[13] – ренин-ангиотензин-альдостероновой системы (рис. 7). РААС отвечает за регуляцию кровяного давления и распределения крови в организме человека. В ее работе задействованы почки, печень, сердечно-сосудистая система.
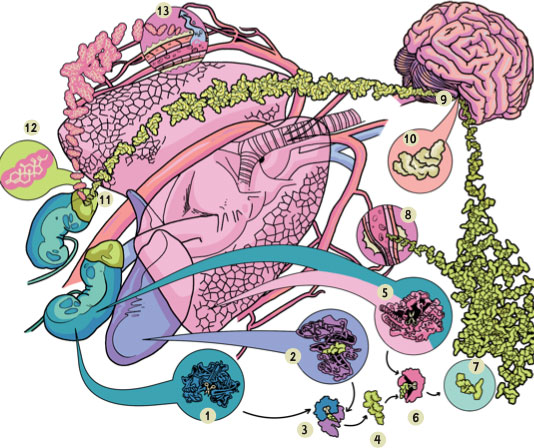
Рисунок 7. Схема работы ренин-ангиотензин-альдостероновой системы:
1 – секреция ренина почками в ответ на снижение внутрипочечного давления; 2 – секреция ангиотензиногена печенью; 3 – отщепление ренином декапептида от ангиотензиногена, высвобождение ангиотензина I; 4 – ангиотензин I; 5 – синтез ангиотензинпревращающего фермента (АПФ) в легких и почках; 6 – преобразование ангиотензина I в ангиотензин II под воздействием АПФ, который отщепляет от него две последние аминокислоты; 7 – ангиотензин II; 8 – сосудосуживающая активность ангиотензина II, за счет которой растет центральное давление; 9 – стимуляция гипофиза ангиотензином II, в результате чего происходит секреция антидиуретического гормона (АДГ); 10 – АДГ; 11 – стимуляция АДГ выброса альдостерона надпочечниками; 12 – альдостерон. 13 – альдостерон задерживает воду и повышает проницаемость мембран клеток, за счет чего увеличивается объем циркулирующей крови и растет кровяное давление
Избыточный синтез одного из участников цепочки РААС в печени (ангиотензиногена), к примеру, приводит к повышенной жесткости тканей сердца – в результате чрезмерного синтеза белков матрикса фибробластами. В итоге работа сердца сильно страдает73, 74, 75, 76, 77. Это происходит следующим образом: активация рецептора к ангиотензину II AT1, который представлен на поверхности клеток многих тканей (сердце, почки, нервная система и др.), вызывает избыточные ответы в кардиомиоцитах и синтез белка внеклеточного матрикса фибробластами сердца.
В фибробластах сердца ангиотензин II повышает синтез коллагена, фибронектина, ламинина и остеопонтина. Гладкомышечные клетки сосудов, стимулированные ангиотензином II, демонстрируют увеличение количества мРНК коллагена, фибронектина, ламинина и тенасцина.
Решение вышеописанных проблем уже проглядывается: опыты на животных показали, что фармакологическое ингибирование компонентов РААС приостанавливает разрастание компонентов матрикса, приводящее к фиброзу, а значит, и патологические процессы в тканях сердца78.
В почках ангиотензин II стимулирует синтез коллагенов, фибронектина и ламинина мезангиальными клетками78, 79.
Способность ангиотензина II стимулировать продукцию TGF-β[14], одного из главных профиброзных факторов, связывают с развитием возрастной сосудистой гипертрофии.
Постоянная активация рецепторов TGF-β приводит к аномальному накоплению соединительной ткани в почках и сосудах, ведущему к фиброзным патологиям80, 81, 82.
Кроме того, продукция TGF-β может индуцироваться при увеличении количества рецепторов RAGE[15]83. Повышение экспрессии этого цитокина может быть прямым следствием процессов гликирования. Возможно, если получится изобрести препарат, «отменяющий» гликирование, вред от избытка TGF-β тоже снизится84, 85, 86.
Таким образом, жесткость почки (за счет синтеза коллагена) растет как напрямую от воздействия ангиотензина II, так и в результате продукции TGF-β. Также запускается очередной порочный круг (на этот раз не только в почках, но и в сосудах): от роста количества КПГ (которые для матрикса и есть сшивки) растет уровень TGF-β, который еще сильнее ускоряет разрастание соединительной ткани.
Митохондрии и матрикс – продолжение истории
Митохондрии находятся внутри клетки, матрикс – снаружи. Поэтому работа митохондрий и матрикса тесно связана друг с другом посредством цитоскелета.
Для обновления белков основных структур цитоскелета (микрофиламенты, промежуточные филаменты, микротрубочки) требуется энергия аденозинтрифосфата (АТФ). Поэтому митохондрии в клетке постоянно двигаются, собираясь в местах, где высока потребность в АТФ94. В свою очередь, правильная организация скелета клетки важна для нормального функционирования митохондрий – они тесно взаимодействуют с ним, чтобы поддерживать свою морфологию. Ангиотензин II, о котором мы говорили выше, нарушает нормальную организацию цитоскелетных филаментов, что негативно сказывается на работе митохондрий94.
Как же взаимосвязаны изменение матрикса, нарушение работы цитоскелета и митохондрии?
Ангиотензин II вызывает изменения в процессе синтеза компонентов межклеточного матрикса, что влияет и на цитоскелет клетки. Известно, что если эндотелиальные клетки окружены более жестким матриксом, то их микротрубочки образуются дольше, правда, получаются в итоге более прочными. Клетки, находящиеся на менее жестком матриксе, образуют не такие прочные микротрубочки, но и растут они быстрее95. Микротрубочки, в свою очередь, очень важны для функционирования митохондрий: при их «разборке» митохондрии теряют свою подвижность. Взаимодействие митохондрий с более прочными трубочками ухудшает их структуру и нарушает энергетическую систему в клетке96.
Еще один из механизмов нарушения цитоскелета клетки, помимо действия ангиотензина II, – регуляция уже упомянутым цитокином TGF-β[16] реорганизации актиновых филаментов. Вызывая перегруппировки актиновых филаментов, он влияет на рост и дифференцировку клеток, так как в ядре запускается действие определенных транскрипционных факторов84, 85, 86 (рис. 8).
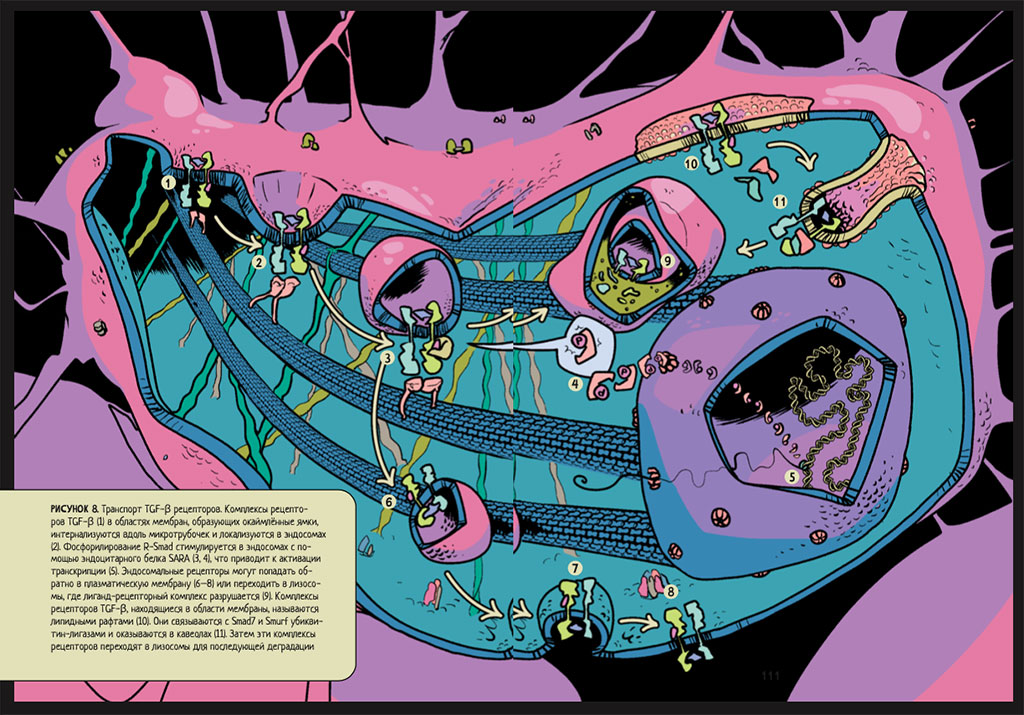
Рисунок 8. Транспорт TGF-β рецепторов. Комплексы рецепторов TGF-β (1) в областях мембран, образующих окаймленные ямки, интернализуются вдоль микротрубочек и локализуются в эндосомах (2). Фосфорилирование R-Smad стимулируется в эндосомах с помощью эндоцитарного белка SARA (3, 4), что приводит к активации транскрипции (5). Эндосомальные рецепторы могут попадать обратно в плазматическую мембрану (6–8) или переходить в лизосомы, где лиганд-рецепторный комплекс разрушается (9). Комплексы рецепторов TGF-β, находящиеся в области мембраны, называются липидными рафтами (10). Они связываются с Smad7 и Smurf убиквитин-лигазами и оказываются в кавеолах (11). Затем эти комплексы рецепторов переходят в лизосомы для последующей деградации.
Аиша Мелуан и ее коллеги показали, что белок SPARC[17] влияет одновременно и на изменения состава внеклеточного матрикса, и на функцию митохондрий в мышечных клетках100.
В мышцах SPARC синтезируется при строительстве или заживлении мышечной ткани. Еще он обладает способностью связываться с коллагенами разных типов, за счет чего влияет на перестройку и формирование внеклеточного матрикса102.
Что касается митохондрий, то этот белок влияет на их развитие путем взаимодействия с индуктором биогенеза митохондрий – белком AMPK (Adenosine Monophosphate-activated Protein Kinase, «протеинкиназой, активируемой аденозинмонофосфатом»)103. Таким образом, SPARC, как и ангиотензин II, и TGF-β, может выступать связующим звеном между работой митохондрий и процессами, протекающими в межклеточном матриксе.
Та же группа ученых представила еще один механизм взаимодействия митохондрий с матриксом при помощи SPARC104. Схема их непростых взаимодействий представлена на рисунке 9.
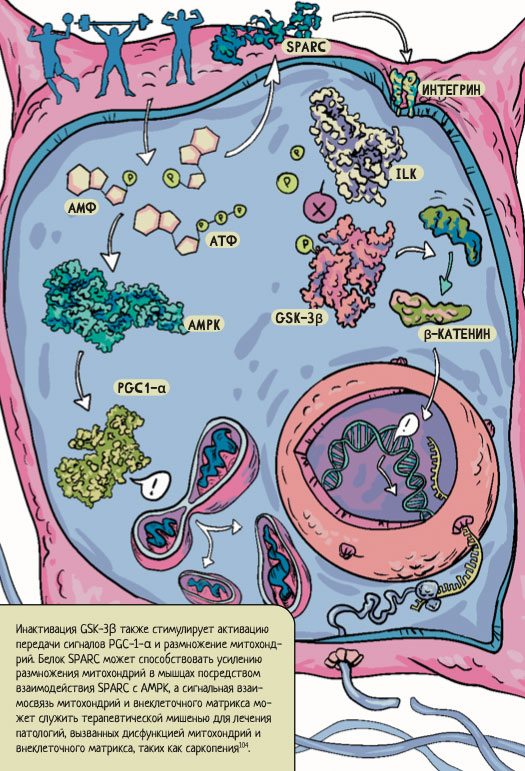
Рисунок 9. Влияние белка SPARC на экспрессию внеклеточного матрикса и на репликацию и транскрипцию митохондриальной ДНК. Физические упражнения вызывают активацию AMPK за счет увеличения соотношения аденозинмонофосфата к аденозинтрифосфату (AMФ/ATФ). Это, в свою очередь, индуцирует биогенез митохондрий посредством активации гамма-рецептора, активируемого пролифератором пероксисом коактиватора-1-альфа (PGC1-α), и повышает уровень экспрессии белка SPARC, что активирует интегрин-связанную киназу (ILK). Последняя фосфорилирует и инактивирует киназу гликогенсинтазы-3-бета (GSK-3ß), что приводит к стабилизации β-катенина и выработке белков мышечного внеклеточного матрикса. Инактивация GSK-3β также стимулирует активацию передачи сигналов PGC-1-α и размножение митохондрий. Белок SPARC может способствовать усилению размножения митохондрий в мышцах посредством взаимодействия SPARC с AMPK, а сигнальная взаимосвязь митохондрий и внеклеточного матрикса может служить терапевтической мишенью для лечения патологий, вызванных дисфункцией митохондрий и внеклеточного матрикса, таких как саркопения104.
В других недавних работах описана взаимосвязь нарушения функций митохондрий и вызванного им избытка кальция с нарушением структуры внеклеточного матрикса в мышцах104, 105. Общая цепь событий в данном случае может быть представлена таким образом: повышенная продукция ангиотензина II в организме вызывает дисфункцию митохондрий и окислительный стресс, что приводит к нарушению гомеостаза кальция. Это, в свою очередь, ведет к нарушению структуры внеклеточного матрикса и дистрофии мышц, а следовательно, к саркопении.
Перекисное окисление липидов
Перекисное окисление липидов (ПОЛ) происходит в первую очередь во внутренней мембране митохондрий, которая находится в активном контакте со свободными радикалами, а также в клеточной мембране нейронов. Этот процесс изменяет физические свойства мембран, их текучесть и работу электрон-транспортной цепи митохондрий. Продукты ПОЛ токсичны и повреждают некоторые важнейшие долгоживущие молекулы: гистоны, белки ядерных пор, структурные белки и ДНК106.
По своей сути ПОЛ – это повреждение полиненасыщенных жирных кислот свободными радикалами, которое происходит в митохондриальных и клеточных мембранах. Двойные связи полиненасыщенных жирных кислот особенно чувствительны к воздействию свободных радикалов, так как легко «разрываются», присоединяя их. В результате этого образуются диальдегиды, пероксиды и другие продукты окисления. Все это запускает цепную реакцию окисления липидов.
Ряд исследований указывает на количественную связь между процессом ПОЛ и образованием поперечных сшивок белков матрикса107. Так, например, один из продуктов ПОЛ, малондиальдегид, образует такое же количество сшивок с белками, как и глюкоза, что было показано в экспериментах in vitro107. Из всего этого следует, что процесс окисления жирных кислот отрицательно влияет на состояние внеклеточного матрикса, что, как мы уже знаем, приводит к неприятным последствиям: старению тканей, их фиброзу и нарушению их функций.
В клетках долгоживущих видов животных меньше полиненасыщенных жирных кислот108, 109, 110. В связи с этим можно предположить, что они не только меньше страдают от ПОЛ, но и процесс изменения белков внеклеточного матрикса у них идет медленнее за счет снижения реакционной способности жирных кислот110.
Существуют способы замедлить скорость перекисного окисления жирных кислот. Так, уже более десяти лет коллектив российского исследователя Михаила Щепинова разрабатывает подход для продления жизни и лечения ряда заболеваний, вызванных избыточным синтезом свободных радикалов. Они используют модифицированные жирные кислоты. Водород в них заменен на дейтерий (изотоп, имеющий больший атомный вес и более прочную связь с атомом углерода). Измененные жирные кислоты более устойчивы к окислению и предотвращают разрушение клеточной мембраны111, 112, 113, 114.
Как же быть?
Теперь поговорим о том, что же можно сделать, чтобы замедлить процесс старения внеклеточного матрикса и решить проблемы, с ним связанные.
Посмотрим на то, как ученые ищут пути решения одной из самых актуальных проблем, связанных с возрастным ремоделированием матрикса, – проблемы гликирования. Уже идет разработка веществ, которые могли бы ингибировать этот процесс или поворачивать его вспять. Рассмотрим некоторые из них.
Растворимая форма RAGE
Известно, что в ответ на стимуляцию рецепторов RAGE конечными продуктами гликирования и возникающий вследствие этого воспалительный стресс клетки нашего организма вырабатывают растворимую форму RAGE. Она служит биомаркером индуцированного КПГ воспаления115, 116.
Также она способна конкурировать за связывание КПГ с рецептором RAGE на поверхностях мембран и таким образом замедлять процесс воспаления. Конечно, природная форма растворимого рецептора RAGE плохо подходит для применения в качестве лекарства из-за ее короткого периода полураспада, большой молекулярной массы и других недостатков, свойственных природным белкам.
Японские исследователи разработали гибридные белковые «ловушки»117 для КПГ, состоящие из участка рецептора RAGE, связывающего КПГ, и эластиноподобного белка, формирующего устойчивые коацерваты. У подобных продуктов белковой инженерии уже есть будущее в качестве противовоспалительных, ранозаживляющих и геропротекторных лекарств.
С той же целью можно использовать и небелковые агенты с меньшей молекулярной массой. Например, ДНК-аптамеры – олигонуклеотиды, нацеленные на блокаду рецепторов RAGE, связывающихся с КПГ белков матрикса. Их планируют использовать для восстановления поврежденных из-за гликирования тканей органов118.
Способность блокировать RAGE-рецепторы обнаружена у некоторых искусственно созданных аминокислот119, а также у дейтерированных полиненасыщенных жирных кислот120, хебуловой кислоты121, кверцетина122 и молекулы GLY-230123.
Главный недостаток этого подхода в том, что в низких концентрациях блокаторы КПГ неэффективны, а в высоких – токсичны. Систему, однако, можно изменять, подбирая комбинации компонентов. В результате успешного подбора блокаторы будут действовать так же эффективно, но из-за более низких концентраций окажутся менее ядовиты.