
Миелопролиферативные новообразования
82. Выбор терапии первой линии хронического миелолейкоза: моделирование клинико-экономических факторов / В. А. Шуваев, К. М. Абдулкадыров, И. С. Мартынкевич, М. С. Фоминых // Клиническая онкогематология. Фундаментальные исследования и клиническая практика. – 2015. – Т. 8, № 1. – С. 78–83.
83. Итоги 12-летней терапии ингибиторами тирозинкиназ больных в поздней хронической фазе хронического миелолейкоза после неудачи лечения ИФН-а / О. В. Лазарева, А. Г. Туркина, Г. А. Гусарова и др. // Сибирский научный медицинский журнал. Бюллетень СО РАМН. – 2015. – Т. 35, № 1. – С. 90–97.
Глава III. Классические Ph-негативные миелопролиферативные новообразования – общая характеристика и общность патогенеза
Термин «классические» Ph-негативные миелопролиферативные новообразования не входит в настоящее время в классификацию МПН, однако широко используется и берёт начало ещё со статьи W. Dameshek в которой МПН, известные на то время (хронический миелолейкоз, истинная полицитемия, эссенциальная тромбоцитемия, первичный миелофиброз), впервые были объединены на основании общности патогенеза и клинических проявлений [1]. После открытия филадельфийской хромосомы и участия белка BCR::ABL в патогенезе заболевания из этой группы был выделен ХМЛ, а остальные новообразования (ИП, ЭТ, ПМФ) стали называться Ph-негативными МПН. Общность их патогенеза получила дополнительные доказательства после открытия патогенетической роли JAK-STAT сигнального пути [2, 3].
Причина возникновения Ph-МПН в настоящее время остается неизвестной. Наиболее вероятен комплексный генез возникновения заболевания, когда предрасположенность к болезни реализуется под влиянием внешних факторов, воздействующих на интактный геном и приводящий к малигнизации клетки [24–26]. Наследственная предрасположенность к заболеванию может иметь место при наличии родственников больных миелопролиферативными новообразованиями (МПН). Относительный риск развития ИП у родственников больных МПН составляет 5,7 [27] и может быть ассоциирован с носительством 46/1 гаплотипа гена JAK2 [28].
Патогенетически МПН представляют собой клональный миелопролиферативный процесс, развивающийся в результате злокачественной трансформации в ранних гемопоэтических предшественниках с последующей соматической мутацией в гене янускиназы рецепторов цитокинов. Повышенная пролиферация миелоидных ростков кроветворения, в большей степени эритроидного при ИП, или мегакариоцитарного при ЭТ, постепенно приводит к развитию очагов экстрамедуллярного кроветворения (спленомегалии), что особенно характерно для ПМФ, повышению риска развития сосудистых тромбозов и тромбоэмболий. Длительная пролиферация патологических гемопоэтических клеток сопровождается фиброзом и замещением деятельного костного мозга волокнами коллагена – развитием ретикулинового и коллагенового миелофиброза, в последней части своего развития, завершающегося остеосклерозом. У части больных накопление повреждений в геноме и дальнейшее прогрессирование болезни завершается фазой бластной трансформации. Одним из ключевых моментов патогенеза МПН считается активация JAK-STAT сигнального пути, обусловленная наличием мутации в гене янускиназы рецепторов цитокинов JAK2 в 617 положении, приводящая к замене фенилаланина на валин – JAK2V617F [4–7], мутациями в генах кальретикулина (CALR) [8, 9], рецептора тромбопоэтина (MPL) [10, 11] или более редко в 12 экзоне JAK2 [30, 31], еще реже наблюдается активация JAK-STAT сигнального пути, связанная с потерей торможения фосфорилирования янускиназ из-за мутации в гене LNK белка SH2B3, между кодонами 208 и 234 [12], или мутациями в генах семейства супрессоров сигнала цитокинов SOC, наиболее часто SOC3 [13] или гиперметилирования CpG участков в генах SOC1 и SOC3 [14]. В последующем могут присоединяться и мутации в других генах: EZH2 [15] и TET2 [16], включающие эпигенетические механизмы.
В настоящее время нет четкого объяснения развития при активации одного и того же сигнального пути JAK-STAT различных нозологических форм: истинной полицитемии (ИП), первичного миелофиброза (ПМФ) или эссенциальной тромбоцитемии (ЭТ). Для объяснения данного феномена предложено несколько патогенетических гипотез:
• носители мутаций – различные стволовые клетки при разных заболеваниях;
• различный уровень активности мутантного JAK2V617F обусловливает особый фенотип заболевания – теория мутационной нагрузки;
• специфический генотип больного – наследственная предрасположенность;
• молекулярные события, предшествующие возникновению мутации в гене JAK2;
• вклад немутационных факторов – эпигенетические механизмы, патологическая экспрессия микроРНК и др. [17, 18].
Janus-киназа является представителем семейства нерецепторных тирозинкиназ. Мутация вызывает замену 1849 нуклеотида G→T, которая в свою очередь приводит к замене в 14 экзоне гена JAK2 фенилаланина на валин в кодоне 617. Молекулы содержат около 1100 аминокислот с общей массой 120–140кДа. Структурно они состоят из семи гомологичных участков, формирующих четыре домена: киназный (JH1), псевдокиназный (JH2), домен с гомологией Sarc онкобелка (SH2), FERM домен. Первый с углеводного окончания молекулы домен (JH1) является типичной тирозинкиназой с каталитической активностью и очень схож с каталитическим доменом тирозинкиназ эпидермального ростового фактора. Следующий домен (JH2) структурно похож на тирозинкиназный домен, но лишен каталитической активности и выполняет регуляторные функции активности [19]. Эта особенность в виде двух похожих участков дала название всему семейству, посвященное древнеримскому богу Янусу, имевшему два лица. SH2 домен облегчает связывание других белков с JAK, домен FERM, расположенный с аминокислотного окончания молекулы и взаимодействует с трансмембранными белками – рецепторами некоторых цитокинов, регулируя активность JAK-киназы [20, 21].
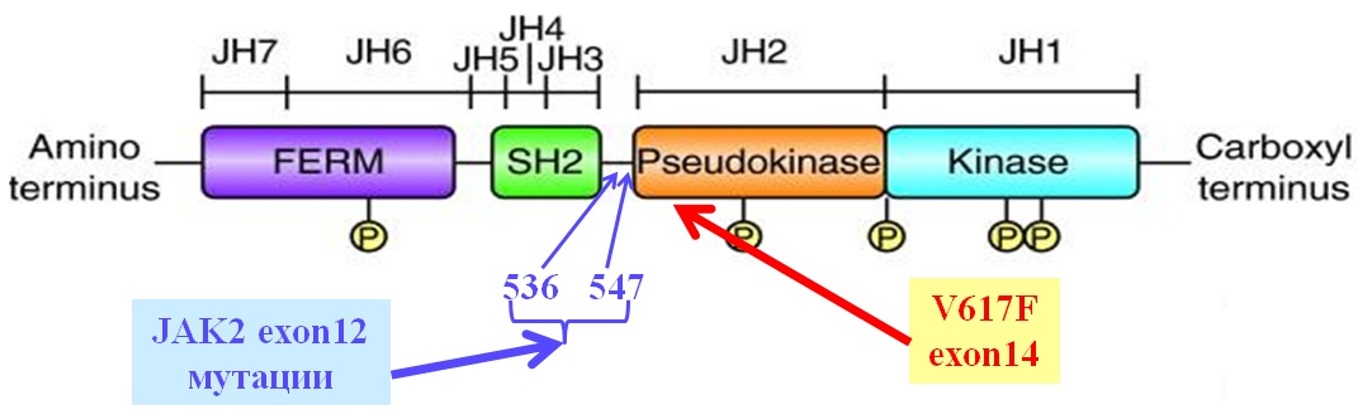
Рисунок III-1. Структура гена JAK2 и место точечных мутаций, обусловливающих независимую активацию [20].
Впервые в эволюционном отношении Janus-киназы возникают у примитивных хордовых. У млекопитающих семейство Janus-киназ представлено четырьмя белками: JAK1, JAK2, JAK3 и TYK2. В настоящее время JAK2V617F мутация описана не только при ИП, но и при других миелоидных новообразованиях. Однако она никогда не определялась у пациентов с опухолями лимфатической ткани, эпителиальными опухолями и саркомами [22]. Локализация генов, кодирующих соответствующие белки и участие в сигнальных путях конкретных цитокинов приведены в таблице III-1.
Таблица III-1. Локализация генов и сигнальные пути цитокинов с участием Janus-киназ [23]

На клеточном уровне Janus-киназы располагаются в цитозоле и локализованы рядом с эндосомами и клеточной мембраной вблизи цитокиновых рецепторов. Белки семейства Janus-киназ участвуют в регуляции многих процессов. Одним из наиболее значимых является передача цитокинового сигнала в ядро с целью стимуляции пролиферации посредством JAK-STAT сигнального пути, схематично представленного на рисунке III-2. При активации цитокинового рецептора происходит изменение его конформационной структуры, которое вызывает ауто- и/или трансфосфорилирование двух JAK-киназ. Janus-киназы, в свою очередь, фосфорилируют внутриклеточную часть цитокинового рецептора. STAT-белки связываются с фосфорилированными частями цитокиновых рецепторов, и также, фосфорилируются Janus-киназами. Связывание STAT-белков с фосфором, позволяет им образовывать активные димеры, которые, проникая в ядро, регулируют экспрессию генов [24]. Такой путь лежит в основе передачи сигнала от рецепторов цитокинов посредством JAK2-киназы в клетках-предшественниках миелопоэза и обусловливают общий патогенез миелопролиферативных новообразований [2]. Одним из ключевых моментов патогенеза часто является возникновение точечной мутации в 1849 положении гена JAK2 в виде замены гуанина на тимин, в результате чего происходит трансформация фенилаланина на валин в кодоне 617 регуляторного домена JH2-псевдокиназы белка JAK2. Это приводит к независимой активации янускиназы и фосфорилированию вторичных мессенджеров в отсутствие стимуляции рецепторов. Данные изменения приводят к активации JAK-STAT сигнального пути и увеличению пролиферации миелоидного ростка.
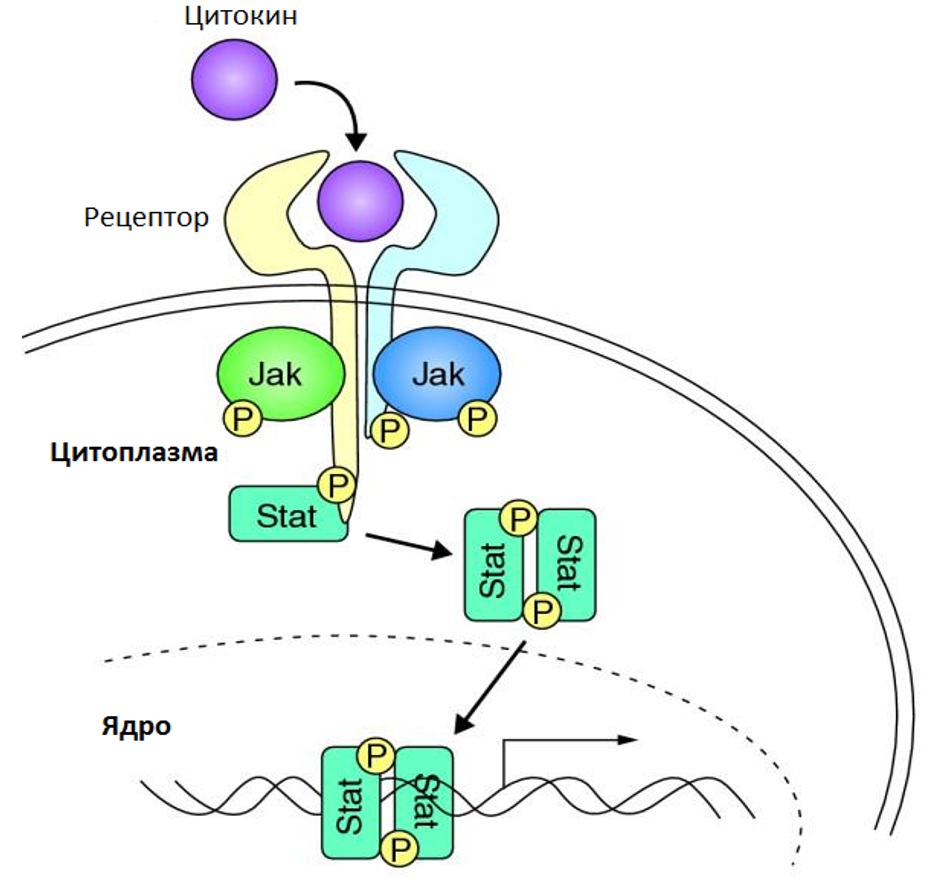
Рисунок III-2. Схема JAK-STAT сигнального пути [23].
Мутация JAK2V617F обнаруживается в полипотентных стволовых клетках – общих предшественниках миело- и лимфопоэза, однако для активации пролиферации посредством JAK-STAT сигнального пути требуется совместная экспрессия с рецепторами цитокинов I типа: эритропоэтина, гранулоцитарного колониестимулирующего фактора и тромбопоэтина. Данный факт является объяснением того, что при наличии JAK2V617F происходит изолированная гиперплазия миелоидного ряда при отсутствии изменений в лимфопоэзе, несмотря на наличие в лимфоидных клетках той же мутации гена JAK2 [45].
При сравнении характеристик JAK2V617F-мутантных клонов у больных истинной полицитемией (ИП), первичным миелофиброзом (ПМФ) и эссенциальной тромбоцитемией (ЭТ) было установлено, что частота гомозиготного носительства JAK2V617F мутаций составляла 30 % при ИП и ПМФ по сравнению с 2–4 % при ЭТ [25]. При этом частота гетерозигот JAK2V617F по данным другого исследования составляет 67,8 % при ИП и 57,6 % при ЭТ [26]. При изучении аллельной нагрузки JAK2V617F количественным ПЦР в реальном времени в группе больных миелопролиферативными новообразованиями (МПН) оказалось, что наименьшая аллельная нагрузка при ЭТ (26±15 %), тогда как у больных ИП (48±26 %), ПМФ (72±24 %), постполицитемическим (пИП-МФ) и посттромбоцитемическим (пЭТ-МФ) (46±30 %) она значительно выше [27]. Полученные результаты легли в основу теории «мутационной нагрузки» развития МПН: различный фенотип нозологического варианта МПН: ИП, ПМФ или ЭТ обусловливается различной степенью аллельной нагрузки JAK2V617F и, в результате, различной активностью функционирования JAK-STAT сигнального пути.
Мутации в генах EZH2 (ген каталитической единицы метилтрансферазы гистонов) и TET2 (TET фермент участвует в превращении 5-метилцитозина в 5-гидроксиметилцитозин), сопутствующие мутациям JAK2 при ИП в 3 % и 16 % случаев соответственно, вносят эпигенетические нарушения в регуляцию транскрипции [15, 28]. Присоединение этих и других (ASXL1, CBL, IDH1/2, IKZF1 и пр.) трансформирующих течение заболевания мутаций может инициировать развитие бластной трансформации (рисунок III-3). Морфологический субстрат заболевания (бласты) при разных вариантах бластного криза после трансформации может содержать или не содержать мутации JAK2 гена.
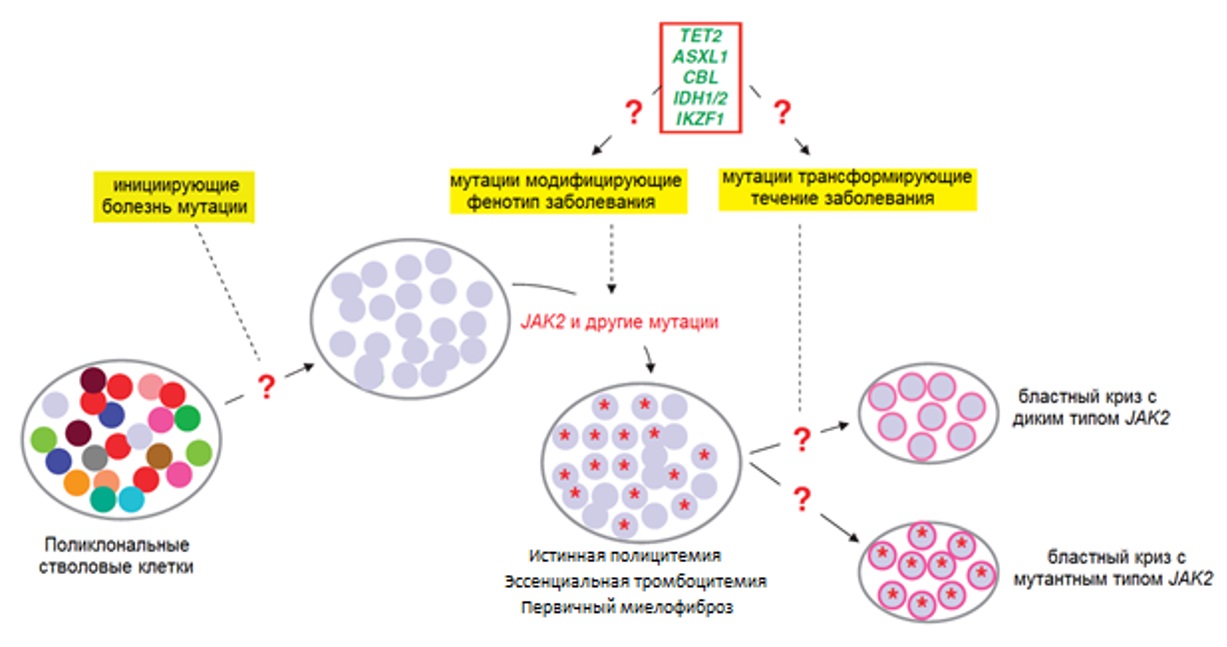
Рисунок III-3. Молекулярно-генетический патогенез миелопролиферативных новообразований [29].
Гиперплазия кроветворения при МПН может сопровождаться патологической выработкой цитокинов, приводящей к вторичному воспалению и изменениям стромы костного мозга. Цитокинами, вовлеченными в этот механизм, являются трансформирующий фактор роста бета миелоидных предшественников (TGF-β), ростовой фактор, вырабатываемый тромбоцитами (PDGFR) и эндотелиальный сосудистый фактор роста (VEGF), которые могут приводить к развитию вторичного миелофиброза, остеосклероза и ангиогенеза [30]. Патологическая выработка цитокинов, хемокинов и металлопротеиназ может участвовать в извращенном межклеточном взаимодействии нейтрофилов, моноцитов и мегакариоцитов, приводя к выходу CD34+ миелоидных предшественников и эндотелиальных клеток в периферическую кровь с развитием очагов экстрамедуллярного кроветворения, в первую очередь миелоидной метаплазии селезенки [31–33]. Результатом длительного влияния этих изменений может быть развитие миелофиброза и остеосклероза.
Подробно по этапам развитие миелофиброза как реакции стромы костного мозга на патологическую секрецию цитокинов представлено на рисунке III-4.
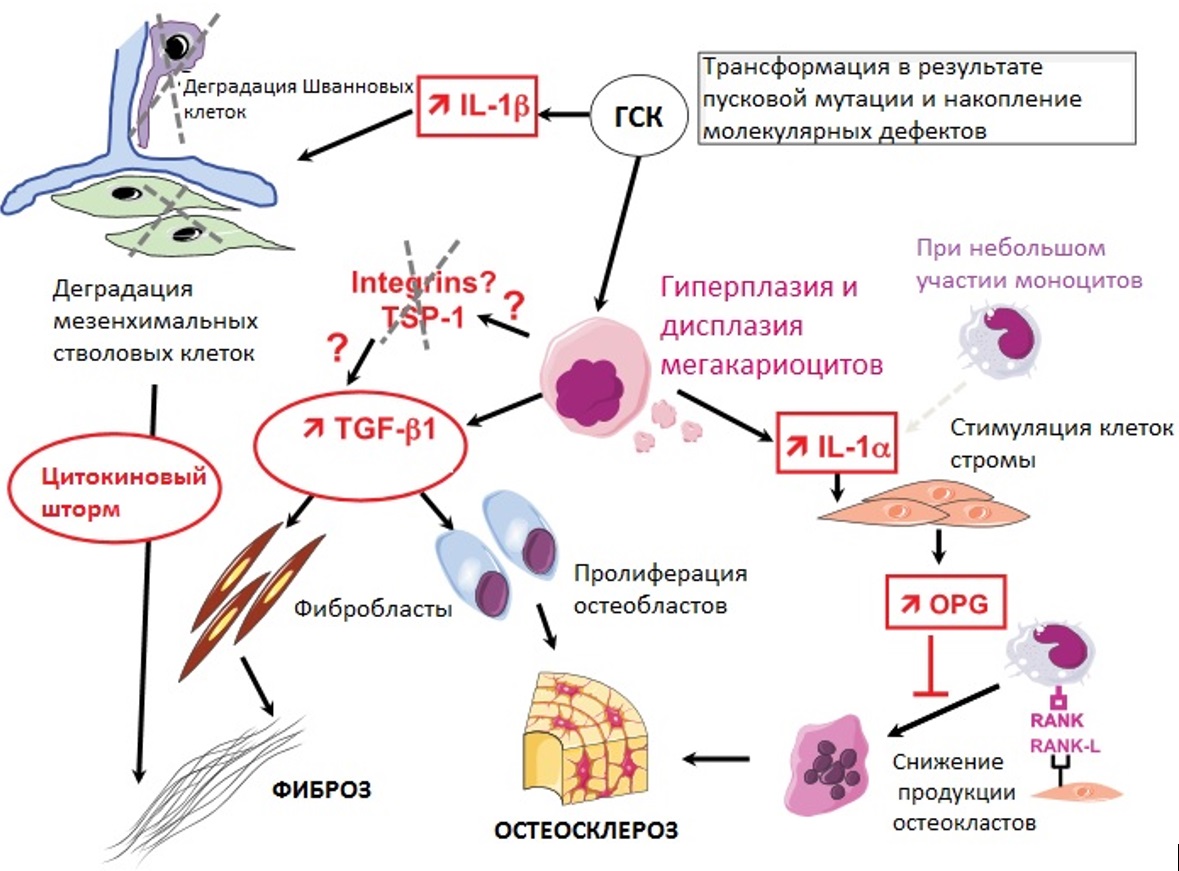
Рисунок III-4. Развитие фиброза костного мозга как реакция стромы костного мозга на аберрантную секрецию цитокинов [34].
Lundberg с соавт. в 2014 г. предложили модель клональной эволюции, объединяющую патогенез всех МПН (рис. III-5) [35]. Инициирующим и единственным событием в патогенезе заболевания примерно у половины больных являются мутации генов JAK2 и CALR. Такие МПН отличаются низким риском прогрессии и бластной трансформации, причем мутации гена CALR ассоциированы с более благоприятным течением заболевания и меньшим риском прогрессии.
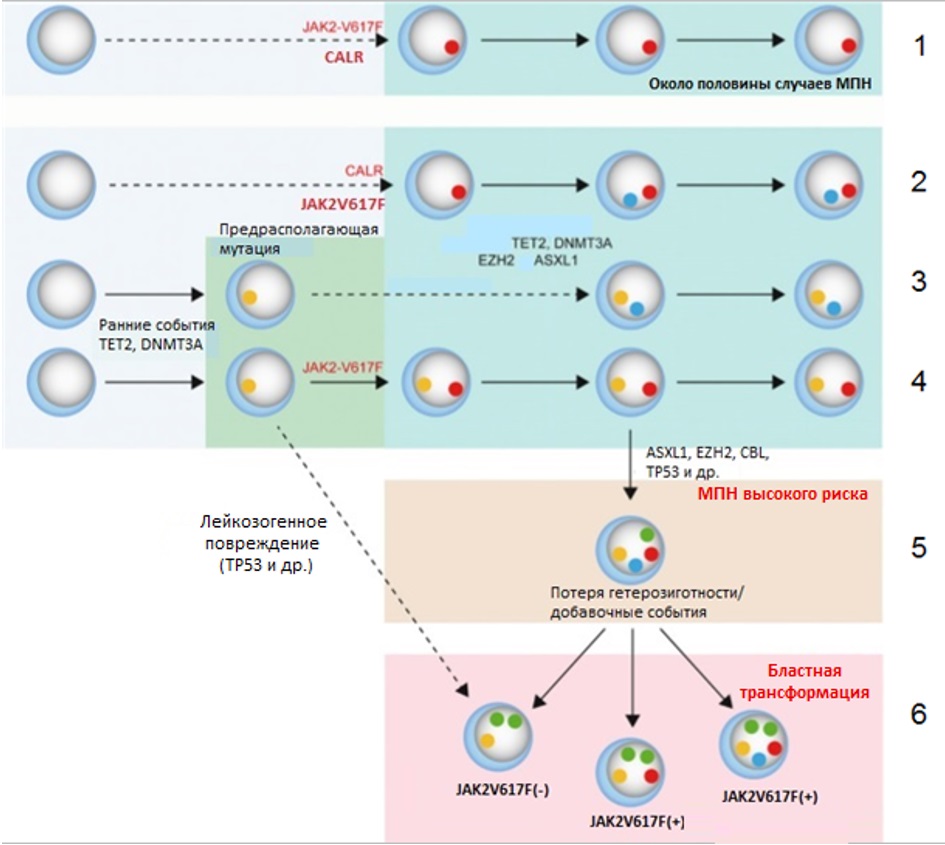
Рисунок III-5 – Общая модель молекулярно-генетического патогенеза МПН [35].
1 – МПН с низким риском прогрессии (ИП, ЭТ); 2 – «продвинутые» формы МПН (ПМФ); 3 – три-негативные МПН; 4 – МПН из клона с повышенным миелопролиферативным потенциалом; 5 – МПН высокого риска с большой мутационной нагрузкой; 6 – бластная трансформация
Полагают, что во всех случаях CALR+ МПН мутация данного гена является самым ранним инициирующим событием. При этом появление дополнительных мутаций (TET2, DNMT3A, EZH2, ASXL1 и др.) приводит к развитию более «продвинутых» форм МПН, таких как ПМФ, и ухудшению прогноза. Пациенты без наличия мутаций в генах JAK2 и CALR могут быть носителями клона, первоначально не являющимся патологическим, но обладающим повышенным пролиферативным потенциалом (вследствие мутаций ТЕТ2, предрасполагающего гаплотипа, неизвестных до настоящего времени мутаций). Появление новых соматических мутаций в генах JAK2, DNMT3A, EZH2, ASXL1 и др. приводит к быстрой манифестации признаков МПН и ухудшению прогноза. Если добавочным событием в клетках, несущих «предрасполагающую» мутацию, будет лейкозогенное повреждение (например, мутация TP53), то возможно развитие ОМЛ de novo без стадии МПН. Бластной трансформации МПН способствует последующее накопление мутаций в генах, участвующих в процессах дифференцировки и регуляции клеточного цикла (EZH2, ASXL1, IDH1/2, CBL, TP53). При этом большинство данных мутаций может обнаруживаться задолго до развития бластного криза. Вероятно, пусковым звеном бластной трансформации при этом будет вовлечение в процесс пока еще не исследованных мутаций или потеря гетерозиготности (например, по ТР53).
Таким образом, патогенез МПН представляет собой последовательный процесс многоступенчатой аккумуляции соматических повреждений, приводящих к нарушениям пролиферации и дифференцировки клеток. Баланс между ними определяет фенотип новообразования (рисунок III-6).
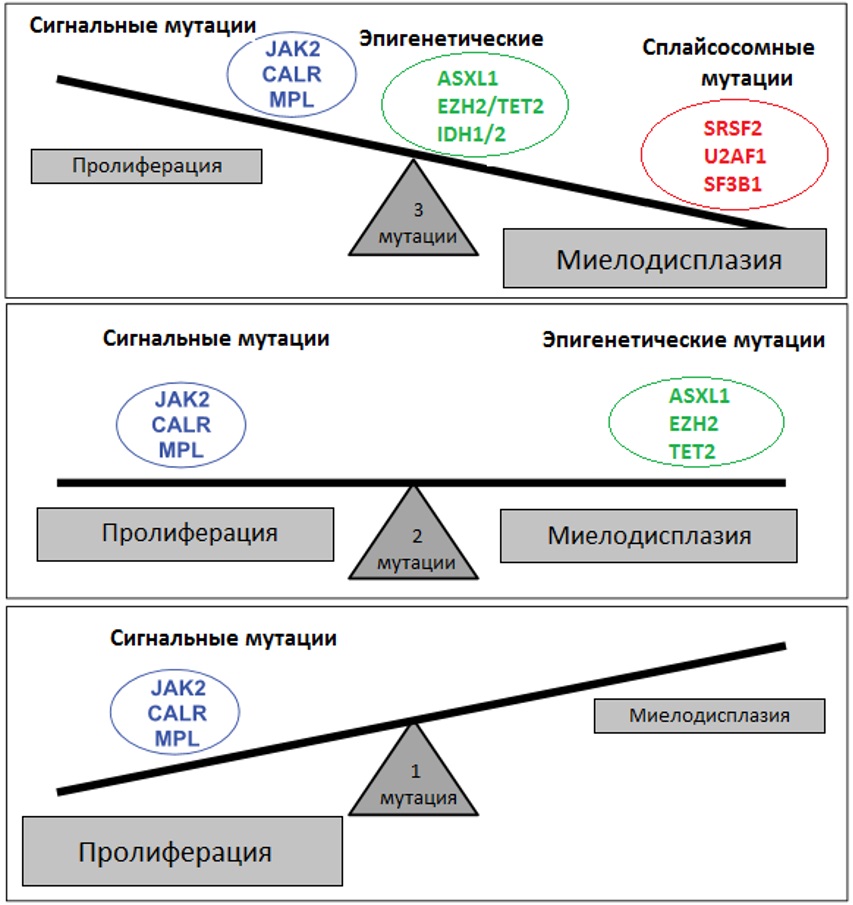
Рисунок III-6. Влияние типа и числа мутаций на фенотип МПН [34].
Достигнутый прогресс в области изучения МПН за последние десятилетия позволил расшифровать основное звено их молекулярного патогенеза. Вместе с тем остается много вопросов связанных с гетерогенностью клинических проявлений, механизмами развития различных нозологических форм при одном молекулярно-генетическом повреждении, необходимостью прогнозирования скорости прогрессирования патологического процесса. Вопросы лечения МПН пока остаются нерешенными полностью. Большинство вариантов терапии носит сдерживающий характер и даже таргетная терапия, направленная на молекулярно-генетические механизмы заболевания, вероятно, всего лишь уменьшает риск осложнений, но не модифицирует течение заболеваний. Единственный радикальный метод лечения – алло-ТКМ для большинства пациентов имеет риски, превышающие возможную пользу увеличения выживаемости. Необходимо активное дальнейшее расширение исследований патогенеза Ph-МПН как для улучшения прогнозирования течения заболевания у каждого пациента, так и для разработки новых препаратов и схем лечения для радикального изменения естественного течения этих болезней для достижения длительной общей и беспрогрессивной выживаемости.
Использованная литература1. Dameshek, W. Editorial: Some Speculations on the Myeloproliferative Syndromes / W. Dameshek // Blood. – 1951. – Vol. 6, № 4. – P. 372–375.
2. Vannucchi, A. M. Guglielmelli, P. Molecular pathophysiology of Philadelphia-negative myeloproliferative disorders: beyond JAK2 and MPL mutations / A. M. Vannucchi, P. Guglielmelli // Haematologica. – 2008. – Vol. 93, № 7. – P. 972–976.
3. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms / A. Tefferi, J. Thiele, A. M. Vannucchi, T. Barbui // Leukemia. – 2014. – Vol. 28, № 7. – P. 1407–1413.
4. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera / C. James, V. Ugo, J.-P. Le Couedic et al. // Nature. – 2005. – Vol. 434, № 7037. – P. 1144–1148.
5. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis / R. L. Levine, M. Wadleigh, J. Cools et al. // Cancer Cell. – 2005. – Vol. 7, № 4. – P. 387–397.
Конец ознакомительного фрагмента.
Текст предоставлен ООО «ЛитРес».
Прочитайте эту книгу целиком, купив полную легальную версию на ЛитРес.
Безопасно оплатить книгу можно банковской картой Visa, MasterCard, Maestro, со счета мобильного телефона, с платежного терминала, в салоне МТС или Связной, через PayPal, WebMoney, Яндекс.Деньги, QIWI Кошелек, бонусными картами или другим удобным Вам способом.
Вы ознакомились с фрагментом книги.
Для бесплатного чтения открыта только часть текста.
Приобретайте полный текст книги у нашего партнера:
Всего 10 форматов