
Внутренние болезни. Том 2
Альтернативным подходом для блокады Fс-рецептора макрофагальномононуклеарной системы у Rh-позитивных больных с ИТ может быть внутривенное или внутримышечное введение анти-Rho иммуноглобулина G в дозах 0,75 – 4,5 мг в течение 1 – 5 дней.
Даназол представляет собой алкилированный в 17-й позиции синтетический андроген с минимальным вирилизирующим эффектом. Клинический опыт показывает, что этот эффект даназола при ИТ во многом зависит от длительности терапии, возраста и пола больных, а также от состояния их селезенки. В частности, женщины реагируют на даназол лучше мужчин. В то же время ранее спленэктомированные больные (50 – 60 %) на даназол практически не реагируют. Иммуномодулирующий эффект даназола при ИТ связывают с уменьшением продукции антитромбоцитарных антител, а также с уменьшением числа Fс-(IgG-) рецепторов на фагоцитирующих тромбоциты клетках. Рекомендуемые начальные дозы даназола должны быть не менее 600 – 800 мг/сут, в то время как для поддержания эффекта достаточно приема 200 мг два раза в неделю.
Положительный эффект á-интерферона при лечении взрослых больных с ИТ связывают с его прямым ингибирующим влиянием на В-клетки. Обычно для получения эффекта он вводится в дозе 3 млн ед. в течение 12 дней. Основные побочные эффекты интерферона – различные аллергические реакции, лихорадка разной степени выраженности, а главное – утяжеление самой тромбоцитопении (из-за его прямого тормозящего влияния на деление костномозговых элементов).
Из других способов терапии ИТ заслуживают внимания: а) ежедневная очистка плазмы от антитромбоцитарных антител; б) удаление антитромбоцитарных антител с помощью плазмафереза со стафилококковым протеином-А; в) использование больших доз аскорбиновой кислоты, вводимых внутривенно; г) применение дапсона; д) внутривенное введение анти-СD20-антитела – мабтеры; е) использование агониста тромбоцитарного рецептора ромипластина. Два последних препарата дороги. Однако положительный опыт использования их при тяжелых, рефрактерных к стандартной терапии формах ИТ, в том числе после аллоТГСК, обнадеживает.
5.6.2. Тромбоцитопатии
Определение. Тромбоцитопатии – это врожденные нарушения тромбоцитарного звена гемостаза, связанные с продукцией костным мозгом неполноценных в отношении адгезии, агрегации или реакции высвобождения (секреции) пластинок. Они наследуются по аутосомно-доминантному или рецессивному типу и имеют однотипную клиническую картину.
Нарушение адгезии тромбоцитов к субэндотелиальным элементам имеет место при болезни Бернара – Сулье. В основе нарушения гемостаза лежит дефект тромбоцитарной мембраны, а именно отсутствие на ней гликопротеина Ib, что приводит к нарушению взаимодействия тромбоцитов с ФВ (см. цв. вкл., рис. 5.14), а через него – с субэндотелиальными структурами сосудистой стенки. Примером заболевания с нарушением фазы первичной агрегации тромбоцитов может быть тромбастения Гланцмана, при которой главную роль в патогенезе нарушений тромбоцитарного звена гемостаза играет дефицит/дефект мембранного гликопротеинового комплекса IIb/IIIa, являющегося рецептором для фибриногена. Взаимодействие последнего с гликопротеином IIb/IIIa и Са2+ необходимо для агрегации тромбоцитов и ретракции кровяного сгустка. В итоге тромбоциты больных тромбастенией Гланцмана не способны реагировать полноценно ни на один из стимуляторов агрегации, в том числе на АДФ, адреналин и коллаген. При этом имеет место также отчетливое снижение ретракции кровяного сгустка.
Клиническая картина. Геморрагические проявления при наследственных тромбоцитопатиях появляются в раннем детстве и имеют однотипный характер. Доминируют петехии и пятнистые кровоизлияния на коже. Они появляются спонтанно или после небольших травм. Характерны также десневые, носовые и маточные кровотечения. Реже встречаются желудочно-кишечные кровотечения и кровотечения во время операций и родов.
Диагноз. Мысль о врожденной тромбоцитопатии возникает у врача при наличии в клинике кровоточивости капиллярного типа, наличии удлиненной кровоточивости на фоне нормального содержания тромбоцитов в крови. Лабораторное обследование позволяет выявить первичное нарушение адгезии, агрегации или секреции в тромбоцитах, которое может сопровождаться также нарушением ретракции кровяного сгустка.
Дифференциальный диагноз тромбоцитопатий следует проводить с другими геморрагическими диатезами, проявляющими себя нарушением функции тромбоцитов, в частности с болезнью Виллебранда. В отличие от тромбастении Гланцмана, при которой нарушены все виды агрегации тромбоцитов, приболезни Виллебранда прежде всего нарушается реакция на ристомицин (ристоцитин), которая корригируется добавлением ФВ. В отличие же от болезни Виллебранда при синдроме Бернара – Сулье добавление ФВ не корригирует выявленный дефект нарушенной агрегации тромбоцитов на ристоцитин, поскольку он связан с отсутствием на тромбоцитах Ib гликопротеинового рецептора для ФВ. Кроме того, при болезни Виллебранда, помимо нарушения в адгезии тромбоцитов, наблюдается снижение коагуляционной активности фактора VIII (VIII: C), поскольку в норме ФВ выполняет роль носителя коагуляционного компонента фактора VIII и защищает его от разрушения в крови. Наконец, при синдроме Бернара – Сулье в мазках крови могут присутствовать гигантские тромбоциты, что для болезни Виллебранда не характерно.
Лечение. Некоторое улучшение функциональной активности тромбоцитов при кровотечении может быть достигнуто с помощью дицинона (внутривенно, per os) или 1 % раствора АТФ (по 1,0 г внутримышечно ежедневно) со жженой магнезией по 0,5 г 3 раза в сутки в течение месяца или внутримышечно 10 инъекций адроксона (по 1 мл 2 – 4 раза в день). Положительный эффект приносит фитотерапия (настои крапивы, тысячелистника и подорожника).
Прогноз при наследственных тромбоцитопатиях, как правило, благоприятный. Естественно, он ухудшается при кровоизлияниях в мозг и в сетчатку глаз, приводящих к церебральным нарушениям и к потере зрения.
5.6.3. Вазопатии
Геморрагические диатезы, связанные с поражением сосудов, могут иметь врожденный и приобретенный характер.
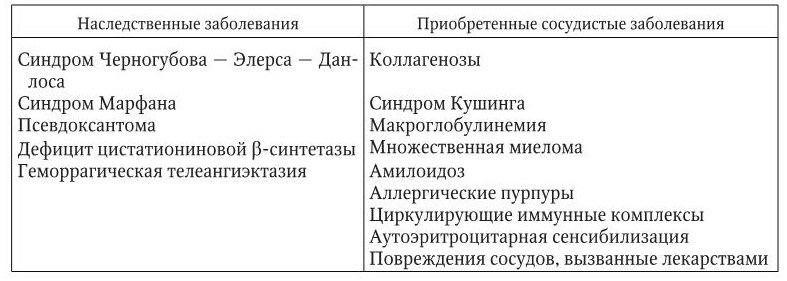
Наследственные геморрагические вазопатии включают болезнь Ослера, синдромы Черногубова – Элерса – Данлоса и Марфана, а также гемангиомы. Их патогенез так или иначе связан с неполноценностью, точнее с неправильным развитием соединительной ткани, в том числе субэндотелия сосудов. Кровоточивость обусловлена малой резистентностью и легкой ранимостью сосудистой стенки, в частности в местах телеангиэктазий, а также слабой стимуляцией в этих участках адгезии и агрегации тромбоцитов.
Кровоточивость при наследственных вазопатиях обнаруживает себя в детском возрасте. Речь идет о легко возникающих геморрагиях в кожу, кровотечениях из слизистых оболочек носа, ротовой полости, желудочно-кишечного тракта. Характерны кровотечения из десен во время чистки зубов. Опасны кровоизлияния в мозг и внутренние органы.
Диагноз ставится на основании рано возникшего геморрагического диатеза у больных с возможными дефектами соединительной ткани, проявляющими себя различными пороками развития, но при отсутствии каких-либо выраженных изменений в тромбоцитарном звене гемостаза. В качестве вспомогательного разграничивающего теста может быть использован аспирин, при назначении которого (0,5 г) у больных с вазопатиями геморрагический диатез должен усиливаться, а при тромбоцитопениях нарастает незначительно.
Лечение наследственных вазопатий носит симптоматический характер. Для остановки кровотечения используют гемостатическую губку, фибриноген, прижигание кровоточащих сосудов и т. д.
Прогноз для жизни относительно благоприятный, хотя оснований для беспокойства у таких пациентов в жизни предостаточно.
Приобретенные вазопатии с проявлениями геморрагического диатеза встречаются в клинике значительно чаще врожденных (табл. 5.3), хотя в клиническом плане мало отличаются от них.
Основные лекарства, вызывающие вазопатии: аспирин, йод; изониазид, мепробамат, метилдопа, пиперазин, хинидин, гипотиазид, мышьяк, толбутамид, аллопуринол, резерпин, индометацин, соли золота, фуросемид, эстрогены, дигоксин, хлорпропамид, хинин, сульфаниламиды, левомицетин, варфарин и др.
Диагноз приобретенных вазопатий ставится при наличии в клинике разной степени выраженности поражений сосудов (атеросклероз, диабетическая ангиопатия, системные васкулиты и др.) в отсутствие отмеченных выше нарушений тромбоцитарного звена гемостаза.
Лечение направлено на коррекцию основного заболевания и неспецифическое устранение симптомов геморрагического диатеза.
Таблица 5.3
Заболевания сосудов, проявляющиеся повышенной кровоточивостью
5.6.4. Коагулопатии
Определение. Коагулопатиями называются геморрагические диатезы, связанные с нарушением плазменного звена гемостаза. Они носят или врожденный (болезнь Виллебранда, гемофилии), или приобретенный характер (ДВС-синдром).
Болезнь ВиллебрандаБолезнь Виллебранда является самой распространенной формой среди наследственных геморрагических диатезов. Заболевание передается по аутосомно-доминантному или рецессивному типу и встречается как у мужчин, так и у женщин. Поскольку ФВ необходим для полноценной адгезии и агрегации тромбоцитов и для поддержания антигемофильного глобулина, его недостаток приводит к нарушению как сосудисто-тромбоцитарного, так и плазменного звеньев гемостаза. В свою очередь, наличие двойного дефекта в системе гемостаза приводит к тому, что болезнь Виллебранда, с одной стороны, напоминает тромбоцитопатию, с другой – гемофилию.
Патогенез. Фактор Виллебранда представляет собой гликопротеин с молекулярной массой от 500 до 12 000 – 20 000 кДа (см. цв. вкл., рис. 5.13). Он состоит из однотипных димерных протомеров (субъединиц), которые кодируются геном хромосомы 12, и синтезируется в клеточных структурах эндотелия и мегакариоцитах. Синтезированный в мегакариоцитах ФВ поступает в тромбоциты, хранится в á-гранулах и выбрасывается из последних под влиянием вызывающих агрегацию стимулов. Роль ФВ в гемостазе связана с его мультимерным строением и наличием в каждой из его субъединиц строго дифференцированных доменов связывания с рецептором тромбоцитарной мембраны, коллагеном, фактором VIII и гепариноподобными гликозаминогликанами. В итоге он функционирует как белок – носитель фактора VIII и одновременно опосредует адгезию тромбоцитов к субэндотелиальным структурам поврежденных сосудов. При этом ФВ выполняет роль моста, соединяющего рецепторы тромбоцитарной мембраны Ib и IIb/VIIIa (см. цв. вкл., рис. 5.13 и 5.14), а с другой стороны – коллаген, миофибриллы эластина, гепариноподобный гликозаминогликан. Нарушение путей синтеза и секреции ФВ приводит к возникновению различных форм болезни Виллебранда, различающихся и по тяжести, и по лабораторным проявлениям.
Клиническая картина. Болезнь Виллебранда проявляет себя очень рано. У детей в возрасте от года до 5 лет могут быть подкожные геморрагии и кровотечения из слизистых оболочек полости рта и носа. Причиной повышенной кровоточивости могут быть травмы и различные хирургические вмешательства (тонзиллэктомия, аденоэктомия, экстракции зубов). Наряду с этим бывают и самопроизвольные носовые кровотечения. На фоне вирусных инфекций верхних дыхательных путей последние могут стать профузными и приводить к анемии. При появлении менструаций у девочек они могут продолжаться от недели до месяца и носить характер меноррагий. При тяжелом течении заболевания встречаются спонтанно возникающие желудочно-кишечные кровотечения, апоплексии яичников, кровоизлияния в сетчатку глаза, гемартрозы и даже межмышечные гематомы. Гемартрозы, не столь частые при болезни Виллебранда, обычно поражают коленные, локтевые и голеностопные суставы. При этом клинико-рентгенологическая картина гемартрозов похожа на гемофильные.
В целом клиническое течение болезни отличается большим полиморфизмом клинических симптомов, что в первую очередь связано с различным содержанием и характером структурных изменений ФВ плазмы и тромбоцитов. На основании этих критериев выделяют несколько подвариантов или типов болезни. Первый, наиболее часто встречающийся тип, выделяется на основании уменьшенного содержания в крови структурно неизмененного ФВ и полного набора мультимеров. Тип II может быть охарактеризован нормальным или субнормальным уровнем измененного по структуре плазменного ФВ, в мультимерном наборе которого или отсутствуют высокомолекулярные формы (подтип IIА), или имеет место его повышенная аффинность к Ib-рецептору тромбоцитарной мембраны, что обнаруживает себя ускоренной ристомициновой агрегацией тромбоцитов в плазме больного. Наконец, при самом тяжелом и редко встречаемом типе III ФВ не определяется совсем ни в плазме, ни в тромбоцитах. В течении заболевания отмечаются периоды обострений и ремиссий. При этом в ходе взросления больного тяжесть клинических симптомов ослабевает.
Диагноз. Мысль о болезни Виллебранда возникает у врача при наличии в клинике рано возникающего геморрагического диатеза, обнаруживающего одновременно нарушения и тромбоцитарного, и плазменного звена гемостаза. Диагноз подтверждается следующими лабораторными признаками: 1) увеличение первичной длительности кровотечения; 2) снижение ристомициновой адгезивности и агрегации тромбоцитов, устраняемое добавлением ФВ; 3) удлинение активированного парциального тромбопластинового времени (АПТВ); 4) снижение коагуляционной активности фактора VIII; 5) снижение уровня ФВ в плазме (определяется иммуноферментным методом) или нарушение его мультимерной структуры.
Дифференциальный диагноз болезни Виллебранда прежде всего проводят с гемофилией А. Опыт показывает, что наибольшие диагностические трудности возникают при разграничении легких форм болезней, когда столь характерное для болезни Виллебранда увеличение длительности кровотечения и нарушение адгезии тромбоцитов может быть не представлено. С другой стороны, эти лабораторные признаки могут обнаружиться из-за образования антитромбоцитарных антител на фоне частых переливаний крови у части больных с гемофилиями. В этом случае решающее диагностическое значение будет иметь прямое определение содержания антигена и активности ФВ, которые при гемофилии или не отличаются от нормы, или даже повышены.
Лечение. При легких и умеренных кровотечениях у больных с I типом болезни Виллебранда с нормальным содержанием ФВ в тромбоцитах показано медленное (10 – 20 мин) внутривенное введение аналога естественного антидиуретического гормона (десмопрессина) в дозе 0,3 мкг/кг массы тела в 30 – 50 мл изотонического раствора натрия хлорида. При этом повышение ФВ и антигемофильного глобулина регистрируется уже через 30 мин после начала инфузии и достигает максимума через 1,5 – 2 ч. Он удерживается на таком уровне в течение 12 ч, отчего требует повторного введения. Однако лечить десмопрессином более 3 сут не рекомендуется в связи с опустошением последним гранул хранения ФВ в клетках эндотелия и отсюда – с необходимостью синтеза в них новых порций этого фактора.
Для лечения больных с вариантами заболевания, где ФВ в гранулах хранения отсутствует, используют свежезамороженную плазму или криопреципитат, богатый ФВ. При определении гемостатической дозы препарата руководствуются коррекцией прокоагулянтной активности фактора VIII и первичной длительности кровотечения. Эффективность терапии оценивают клинически – по уменьшению интенсивности и продолжительности кровотечений и рассасыванию кровоизлияний. В случае переливания препаратов крови, содержащих и ФВ, и антигемофильный глобулин, уровень последнего увеличивается пропорционально введенному фактору и поддерживается на хорошем уровне. Необходимость в заместительной терапии антигемофильным глобулином возникает только при профузных и длительных кровотечениях. В этом случае фактор VIII вводится внутривенно до остановки кровотечения каждые 24 ч из расчета 20 ед./кг массы тела. Что касается остановки кровотечений из слизистых оболочек полости рта и носа, для этих целей может быть рекомендовано местное использование и тромбина, и гемостатической губки или марли. Прогноз при различных подвариантах заболевания неодинаков. Он более благоприятен при умеренно выраженной форме болезни Виллебранда. В то же время тяжелое течение болезни, нередко наблюдающееся у больных с III типом болезни Виллебранда, может привести к анкилозу суставов и к тяжелой анемии.
ГемофилииГемофилии образуют группу наследственно обусловленных заболеваний крови, связанных с нарушением плазменного звена гемостаза. По современным классификациям могут быть выделены гемофилии А, В, С, D и Е.
Гемофилия А – наследственный геморрагический диатез, связанный с нарушенной продукцией антигемофильного глобулина или фактора VIII. Поскольку ответственный за этот белок ген локализован на Х-хромосоме и является рецессивным, гемофилией А в основном болеют мужчины. Женщины же, носители гена гемофилии, имеющие вторую Х-хромосому (гетерозиготы), этим заболеванием не страдают. Исключение составляют лишь девочки-гомозиготы, которые родились в семье больного гемофилией отца и матери – переносчика этой дефективной Х-хромосомы. Кроме того, у 10 – 15 % больных с приобретенной в ходе жизни мутацией гена антигемофильного глобулина гемофилия А может иметь и ненаследственный характер.
Патогенез. Нарушение гена, ответственного за синтез антигемофильного глобулина, чаще всего ассоциируется с продукцией функционально неполноценного фактора VIII (гемофилия А+) или, реже, с его количественным дефектом (гемофилия А–). В любом случае снижение активности фактора VIII приводит к резкому замедлению свертывания крови по внутреннему пути. При этом степень дефицита фактора VIII тесно связана с выраженностью геморрагического диатеза, что находит отражение и в классификации болезни. В частности, при тяжелой форме гемофилии А активность фактора VIII не превышает 5 %, при средней тяжести течения она варьирует от 5 до 10 %, а при легкой – превышает 10 %.
Клиническая картина. Основным симптомом в клинической картине являются периодически повторяющиеся эпизоды кровоточивости. Как правило, они регистрируются в раннем возрасте, но могут появиться и позднее 20 лет. Несмотря на рождение с геном гемофилии, новорожденные дети кровоточат редко, в том числе после перерезки пуповины. До полугода склонность к кровотечениям выявляется у немногих, и они могут появиться при надрыве уздечки языка или подрезании ногтей. Прорезывание молочных зубов проходит без кровоточивости. Однако острые края самих молочных зубов могут быть причиной кровотечений из-за прикусов языка, щек и губ. Когда ребенок начинает вставать и пробует ходить, возможность травматизации резко возрастает. Из-за того, что он падает, появляются гематомы туловища и головы. Когда же ребенок начинает активно ходить и бегать, открываются возможности для кровоизлияния в суставы, прежде всего коленные, локтевые и голеностопные. «Спонтанные» кровотечения (вне травматизации), прежде всего, присущи тяжелым формам гемофилии. С другой стороны, оперативные вмешательства, даже экстракция зубов, обычно сопровождаются повышенной кровоточивостью. Поскольку образование первичной тромбоцитарной пробки при гемофилии не нарушено, эти постоперационные кровотечения у больных гемофилией возникают не сразу, а через несколько часов, но из-за отсутствия выпадения фибрина могут продолжаться днями и даже неделями.
Конец ознакомительного фрагмента.
Текст предоставлен ООО «ЛитРес».
Прочитайте эту книгу целиком, купив полную легальную версию на ЛитРес.
Безопасно оплатить книгу можно банковской картой Visa, MasterCard, Maestro, со счета мобильного телефона, с платежного терминала, в салоне МТС или Связной, через PayPal, WebMoney, Яндекс.Деньги, QIWI Кошелек, бонусными картами или другим удобным Вам способом.
Вы ознакомились с фрагментом книги.
Для бесплатного чтения открыта только часть текста.
Приобретайте полный текст книги у нашего партнера:
Всего 10 форматов