
Миелопролиферативные новообразования
17. Interim Analysis of a Pan European Stop Tyrosine Kinase Inhibitor Trial in Chronic Myeloid Leukemia: The EURO-SKI study / F.-X. Mahon, J. Richter, J. Guilhot et al. // Blood. – 2015. – 56th Annual Meeting and Exposition, San Francisco, CA December 6–9, 2014. – P. Abstract 151.
18. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial / F.-X. Mahon, D. Rea, J. Guilhot, F. Guilhot et al. // The Lancet Oncology. – 2010. – Vol. 11, № 11. – P. 1029–1035.
19. Mahon, F.-X. Discontinuation of tyrosine kinase therapy in CML / F.-X. Mahon // Annals of Hematology. – 2015. – Vol. 94, № 2. – P. 187–193.
20. Hughes, T. P., Ross, D. M. Moving treatment-free remission into mainstream clinical practice in CML / T. P. Hughes, D. M. Ross // Blood. – 2016. – Vol. 128, № 1. – P. 17.
21. Vannucchi, A. M., Guglielmelli, P. Molecular pathophysiology of Philadelphia-negative myeloproliferative disorders: beyond JAK2 and MPL mutations / A. M. Vannucchi, P. Guglielmelli // Haematologica. – 2008. – Vol. 93, № 7. – P. 972–976.
22. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis / A. M. Vannucchi, H. M. Kantarjian, J.-J. Kiladjian et al. // Haematologica. – 2015. – Vol. 100, № 9. – P. 1139–1145.
23. Ruxolitinib Efficacy By Hematocrit Control in Patients with Polycythemia Vera: An Analysis of the RESPONSE Trial / S. Verstovsek, J.-J. Kiladjian, R. Mesa et al. // Blood. – 2014. – Vol. 124, № 21. – P. 3201–3201.
24. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms / T. Klampfl, H. Gisslinger, A. S. Harutyunyan et al. // New England Journal of Medicine. – 2013. – Vol. 369, № 25. – P. 2379–2390.
25. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2 / J. Nangalia, C. E. Massie, E. J. Baxter et al. // New England Journal of Medicine. – 2013. – Vol. 369, № 25. – P. 2391–2405.
Глава II. Хронический миелолейкоз
Хронический миелолейкоз (ХМЛ, код по МКБ10 C92.1) – клональное опухолевое заболевание, обусловленное злокачественным перерождением плюрипотентной стволовой клетки, характеризующееся усилением пролиферации гранулоцитарного ростка без потери способности к дифференцировке, ассоциированное с характерной хромосомной (филадельфийской хромосомой) аномалией. Хронический миелолейкоз характеризуется выраженной гиперплазией миелоидной ткани и миелоидной метаплазией кроветворных органов [1].
Этиология, эпидемиология и патогенез. Этиология заболевания не установлена, обсуждается роль различных факторов (ионизирующее излучение, токсины и инфекции), ни один из которых не получил значимого подтверждения в возникновении заболевания [2].
Традиционно представление о ХМЛ, как о редком заболевании без географической или этнической неоднородности. Заболевание встречается у людей любого возраста и обоего пола, однако у детей крайне редко. Пик заболеваемости приходится на 50-летний возраст. В структуре лейкозов занимает пятое место и составляет 20 % от всех форм лейкозов. Заболеваемость ХМЛ составляет 1–1,5 на 100000 населения в год [1, 2]. В Российской Федерации ежегодно регистрируется 0,58 больных на 100000 населения [3]. По результатам изучения данных в Регистре больных хроническим миелолейкозом в Российской Федерации на момент диагностики соотношение мужчин и женщин в Регистре составляет 44:56, медиана возраста пациентов – 49 лет (диапазон 2–94 года). Пик выявления заболевания (46,3 %) приходится на возрастную группу 40–60 лет. Доля пациентов в возрасте до 40 лет составляет 30,4 %, старше 60 лет – 23,3 %. Среди больных моложе 40 лет соотношение по полу примерно равное. После 40 лет преобладают пациенты женского пола [4].
Благодаря внедрению таргетной терапии препаратами – ингибиторами тирозинкиназ, за последние 10 лет наблюдается снижение смертности, и как результат – постоянный рост распространенности с 3,40 в 2005 г. до 6,41 больных ХМЛ на 100000 населения в 2015 г. [5]. В масштабах страны данный факт привел к увеличению общего количества больных в стране до 6995 человек в 2016 г. [4].
Патогенез ХМЛ хорошо изучен. В 1960 г. в г. Филадельфия (США) P. Nowell и D. Hungerford впервые обнаружили у больного ХМЛ хромосомную аномалию – укороченную хромосому 22, впоследствии названную филадельфийской [6]. Это наблюдение в последующем привело к открытию ключевого момента в патогенезе ХМЛ – образования химерного гена BCR::ABL – продукта обмена генетическим материалом между 9 и 22 хромосомами. При этом в результате слияния двух нормальных генов образуется новый ген, продуцирующий патологический белок, имеющий в 1000 раз более высокую тирозинкиназную активность, чем его нормальный предшественник [7].
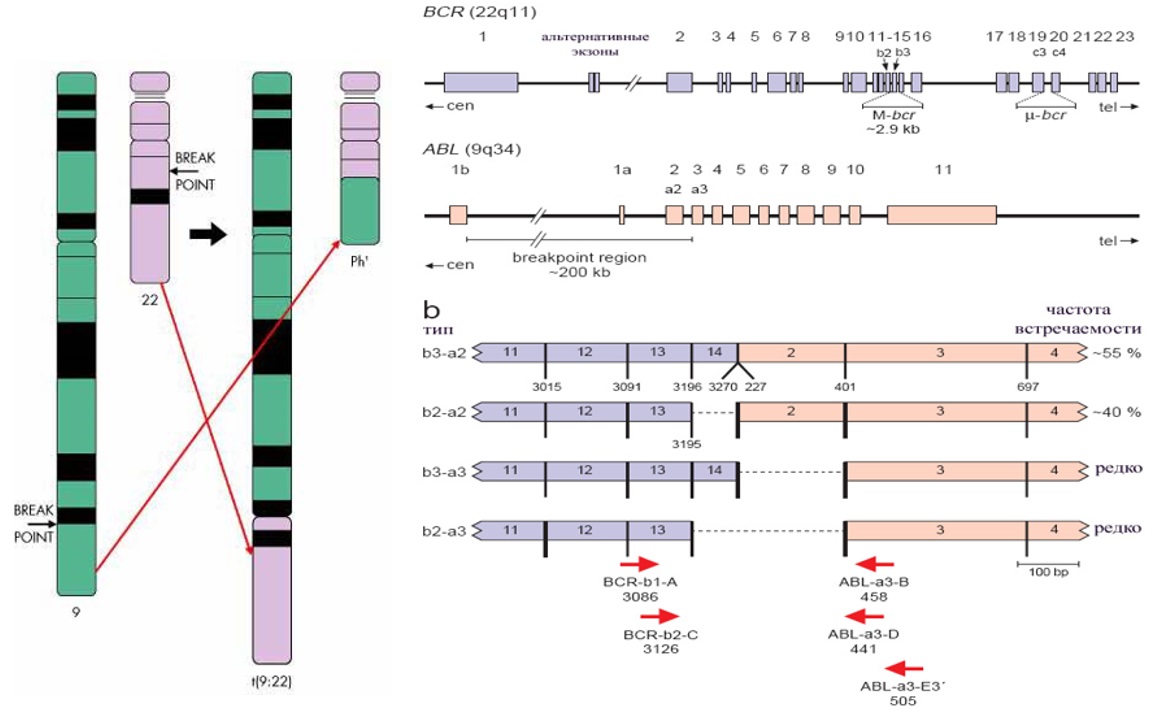
Рисунок II-1. Образование химерного гена BCR::ABL [7].
Роль гена BCR::ABL в патогенезе ХМЛ не ограничивается только повышением размножения клеток. Его повышенная тирозинкиназная активность, придает преимущества лейкемическим клеткам в независимой от влияния сигналов организма пролиферации, блокировании апоптоза – клеточного самоуничтожения, изменения сигнальных путей [8–10].
Клинические проявленияНа начальных стадиях развития патологического процесса повышение пролиферации клеток-предшественников в костном мозге приводит к его гиперплазии и резкому повышению показателей клеток крови, в большей степени нейтрофильных лейкоцитов и тромбоцитов. В настоящее время, благодаря проведению диспансеризации и частому выполнению клинического анализа крови при обращении за медицинской помощью диагноз ХМЛ нередко ставится относительно случайно, без наличия выраженных клинических проявлений. Субъективно в хронической стадии заболевания пациент может не испытывать никаких симптомов или могут присутствовать слабость, потливость, боли в костях, в отдельных случаях – повышение температуры и наличие проявлений геморрагического синдрома (вторичная коагулопатия на фоне резко выраженного гипертромбоцитоза). Размеры печени и селезенки могут варьировать от практически нормальных до резко увеличенных. Картина крови в этом периоде ХМЛ отличается увеличением числа лейкоцитов от умеренного повышения до резко выраженного (400–500 × 109/л). В лейкоцитарной формуле могут определяться все переходные элементы миелоидного ряда, включая промиелоциты и миелобласты. Важной характеристикой формулы является постепенное нарастание количества форм клеток в соответствии с рядом созревания, то есть отсутствие лейкемического провала, присущего острым лейкозам. Реже встречающийся в настоящее время феномен при ХМЛ – это повышение содержания эозинофилов и базофилов (эозинофильно-базофильная ассоциация). Концентрация гемоглобина может быть в пределах нормы или быть сниженным, при этом анемия чаще всего характеризуется как нормоцитарная нормохромная. Количество тромбоцитов может быть разным – в пределах нормы, резко увеличенным или сниженным (тромбоцитопения).
Среди биохимических показателей может отмечаться повышение уровня мочевой кислоты, из-за накопления продуктов пуринового обмена вследствие разрушения избыточной клеточной массы. Данное обстоятельство может приводить к развитию вторичной гиперурикемии и ее клиническим проявлениям (артрит, мочекаменная болезнь, тофусы), вследствие этого бывает, что ХМЛ является случайной находкой при обследовании пациентов по поводу суставной атаки схожей с подагрой.
В пунктате грудины определяется схожая с кровью морфологическая картина. Основную массу клеток в миелограмме составляют клетки нейтрофильного ряда без изменения соотношения в ряду созревания. В костном мозге также как и в крови может отмечаться эозинофильно-базофильная ассоциация.
При гистологическом исследовании трепанобиоптата костного мозга выявляется резко выраженная гиперплазия за счет миелоидных элементов с одновременным уменьшением и даже полным исчезновением жировых клеток. В хронической фазе ХМЛ отмечается полиморфизм клеток с увеличением содержания нейтрофилов различной степени зрелости. Наблюдается также большое количество мегакариоцитов, эозинофильных и базофильных миелоцитов, зрелых эозинофилов и базофилов. Прогрессирование в продвинутые стадии сопровождается нарастанием дисплазии клеточных элементов, увеличением количества бластных форм. При цитогенетическом исследовании у подавляющего большинства больных определяется классическая Ph-хромосома. У 4–5 % больных ХМЛ Ph-хромосома может быть скрытой (не выявляемой при микроскопическом исследовании) или вариантной с участием в траслокации не только 9 и 22 хромосом, но и других хромосом-партнеров. Диагноз ХМЛ в данных случаях может быть верифицирован с помощью FISH исследования с использованием молекулярно-генетических зондов к генам BCR и ABL. Дополнительные хромосомные аберрации (ДХА) за исключением Ph-хромосомы могут наблюдаться у 8–10 % больных в хронической фазе ХМЛ и свидетельствуют о большей генетической нестабильности генома, часть из ДХА являются доказанными факторами высокого риска прогрессирования заболевания. При молекулярно-генетическом исследовании обнаруживается белок – продукт патологического гена BCR::ABL. При ХМЛ могут выявляться свыше 16 различных вариантов белка BCR::ABL. Свыше 95 % больных имеют варианты с разрывом гена BCR в наиболее типичном месте – major breakpoint cluster region (M-bcr): b3a2 или b2a2 с молекулярной массой 210 кДа (p210); менее частые варианты с точкой разрыва в экзоне 1 гена BCR – minor breakpoint cluster region (m-bcr) е1а2 (p190); в экзоне 19 гена BCR – micro breakpoint cluster region (μ-bcr) e19a2 (р230). Редкие варианты транскриптов BCR::ABL образуются при разрыве гена BCR в 6-го по 8-й экзон (variable breakpoint cluster region ν-bcr): e6a2 (p185), e8a2 (p200). Вариабельность транскриптов BCR::ABL также может быть обусловлена нетипичным вовлечением гена ABL в экзоне 3 e13a3 (b2a3, p174), e14a3 (b3a3) [11].
Особенно сложной является дифференциальная диагностика между бластными кризами ХМЛ и острыми Ph+ лимфобластными и миелоидными лейкозами. Для Ph+ ОЛЛ более типичным является вариант белка BCR::ABL с молекулярной массой 190 кДа, однако и при ХМЛ данный вариант транскрипта BCR::ABL не является исключением. Способом проведения дифференциальной диагностики между ХМЛ и острыми Ph+ лейкозами является постановка FISH с молекулярно-генетическими зондами к генам BCR и ABL на нейтрофилах периферической крови. При ХМЛ сохраняется способность к дифференцировке и клетки периферической крови являются потомками трансформированной стволовой клетки и при проведении FISH на нейтрофилах выявляется слитный сигнал зондов BCR и ABL. При Ph+ острых лейкозах имеется блок дифференцировки на уровне бластов и остающиеся зрелые клетки крови происходят из нормального гемопоэза, соответственно результаты FISH с зондами BCR и ABL на нейтрофилах крови отрицательные (слитный ген не определяется) [12, 13].
В настоящее время для оказания помощи пациентам с ХМЛ разработаны и применяются международные рекомендации по диагностике и лечению больных хроническим миелолейкозом. Чаще других используются рекомендации Европейской организации по диагностике и лечению лейкозов (European Leukemia Net – ELN), выпущенные в 2008 [14], 2013 [15] и 2020 гг. Данные рекомендации являются консенсусом широкого круга экспертов, в том числе и с участием российских гематологов (проф. А. Г. Туркина и проф. А. Ю. Зарицкий) [16] и постоянно обновляемые рекомендации Национальной онкологической сети США (National Cancer Comprehensive Network – NCCN) [17]. На основании этих рекомендаций и собственного опыта Национальным гематологическим обществом (НГО) РФ были разработаны отечественные Клинические рекомендации по диагностике и лечению хронического миелолейкоза [18, 19].
Классификация. В течении ХМЛ выделяют 3 фазы, отражающие степень прогрессирования заболевания: хроническую фазу (ХФ), фазу акселерации (ФА), фазу бластной трансформации или бластный криз (БК). Заболевание может быть впервые выявлено на любом этапе течения.
Хроническая фаза (ХФ) является начальной стадией ХМЛ и диагностируется у большинства (более 80 %) впервые выявленных больных. Диагноз ХФ устанавливают при отсутствии признаков фазы акселерации и бластного криза.
Длительная пролиферация опухолевого клона может приводить к накоплению вторичных повреждений генома и переходу в продвинутые стадии заболевания – фазу акселерации (ФА) и бластный криз (БК).
Фазы акселерации и бластного криза ХМЛ характеризуются потерей массы тела больных, присоединением различного рода осложнений (инфекционных, геморрагических, анемического синдрома), повышением температуры, прогрессирующим увеличением размеров селезенки и печени, развитием анемии и тромбоцитопении, нарастанием количества миелобластов в периферической крови. В костном мозге происходит прогрессирующее увеличение количества миелобластов и подавление гемопоэза. В миелограмме и трепанобиоптате отмечается увеличение недифференцированных элементов миелоидного ряда. Вместо полиморфизма клеточного состава в терминальных стадиях ХМЛ обнаруживаются преимущественно миелобласты. Количество зрелых нейтрофилов, а также нормобластов и мегакариоцитов резко уменьшается. При цитогенетическом исследовании в ФА и БК нередко определяются ДХА, при молекулярно-генетическом исследовании в 30–50 % случаев обнаруживаются точечные мутации гена BCR::ABL, обусловливающие резистентность к препаратам таргетной терапии [1, 5, 15, 20, 21].
Фаза акселерации (ФА) определяется у 8–10 % первичных больных ХМЛ и является более продвинутым по сравнению с ХФ этапом развития патологического процесса.
Бластный криз (БК) является наиболее агрессивной стадией ХМЛ. Дебют болезни с бластного криза является неблагоприятным прогностическим признаком и наблюдается у 1–2 % больных ХМЛ.
Для обозначения ФА и БК нередко используется термин «продвинутые фазы заболевания». Медиана продолжительности жизни больных при продвинутых фазах ХМЛ без проведения таргетной терапии ранее составляла 6–12 месяцев [1, 22]. Гепато- и спленомегалия, а также дополнительные хромосомные аберрации (ДХА) не являются критериями продвинутых фаз, согласно современным классификациям.
Фаза ХМЛ оценивается в дебюте заболевания, а также при прогрессировании заболевания, и, обязательно, – при изменении терапии.
Наиболее часто в настоящее время в клинической практике, в том числе и в России используется классификация ELN [19]. Предлагаемые для обсуждения критерии определения фазы акселерации ВОЗ 2016 г. вызывают сомнения, их внедрение также потребует переклассификации в ФА, значительной части случаев ХМЛ, ранее определяемых как ХФ. Критерии различных фаз ХМЛ по классификациям ELN [15] и ВОЗ [23] приведены в табл. II-1.
Таблица II-1. Фазы ХМЛ по классификациям ELN [15] и ВОЗ [23]

В недавно опубликованном пятом пересмотре классификации ВОЗ предлагается полностью исключить из классификации ХМЛ ФА и заменить её на признаки высокого риска прогрессирования. С данным изменением нелегко согласиться, так как прогноз больных в ФА ХМЛ более близок к БК, нежели к ХФ. Таким образом, целесообразнее представляется объединение ФА и БК в общую продвинутую фазу ХМЛ, чем включение больных в ФА к пациентам с ХФ [24].
Диагностика. Диагноз ХМЛ устанавливается на основании данных клинико-лабораторных исследований при обязательном обнаружении Ph-хромосомы и/или химерного гена BCR::ABL [21, 33, 34].
Проведение дифференциального диагноза ХМЛ необходимо прежде всего с заболеваниями и состояниями с реактивным лейкоцитозом (лейкемоидные реакции наиболее часто обусловленные инфекционными и аутоиммунными заболеваниями) и другими миелопролиферативными новообразованиями без наличия гена BCR::ABL: Ph-негативными МПН – первичным миелофиброзом (ПМФ), истинной полицитемией (ИП), эссенциальной тромбоцитемией (ЭТ).
Рекомендации ELN 2020 предлагают следующий объем обследования для установления диагноза ХМЛ:
• физикальное обследование с оценкой размеров селезенки и печени;
• клинический анализ крови с подсчетом лейкоцитарной формулы;
• аспирационная биопсия костного мозга с подсчетом миелограммы; трепанобиопсия с гистологическим исследованием костного мозга при «сухом проколе»;
• цитогенетическое исследование (кариотипирование) клеток костного мозга;
• FISH с зондами на гены BCR и ABL только в случае Ph-негативности;
• качественная ОТ-ПЦР для определения наличия и типа транскрипта гена BCR::ABL;
• электрокардиография;
• стандартная биохимическая панель с серологическим исследованием на гепатит B.
В стандартную биохимическую панель (не расшифрованную полностью в тексте рекомендаций), кроме холестерина, липазы, гемоглобина А1, на наш взгляд целесообразно включать общий билирубин, АСТ, АЛТ, ЛДГ, липиды крови, мочевую кислоту, мочевину, креатинин, общий белок, альбумин, щелочную фосфатазу, электролиты (калий, натрий, кальций, фосфор, магний), амилазу, глюкозу. Также очень важным является сбор анамнеза о наличии сопутствующих заболеваний, степени их активности и приеме лекарственных препаратов. Данная информация имеет значение для выбора ИТК и коррекции доз с учетом межлекарственного взаимодействия.
Количественное определение уровня экспрессии типичных транскриптов гена BCR::ABL по международной шкале не вошло в перечень обязательных исследований при диагностике ХМЛ. Вместе с тем существует ряд исследований показывающих большую прогностическую роль индивидуальной динамики количественного уровня BCR::ABL в течение первых трех месяцев терапии ИТК [25–27]. Таким образом, количественное измерение уровня BCR::ABL при первичной диагностике, хоть и не является обязательным, но может оказаться полезным при последующем мониторинге ответа на терапию ИТК.
Группа риска ХМЛ – понятие, применимое только для ХФ ХМЛ. Группа риска в этой фазе оценивается только на момент диагностики заболевания, до начала терапии. Она рассчитывается на основании прогностически значимых характеристик: низкий, промежуточный, либо высокий риск.
Совокупность критериев, характеризующих группы риска по системам J. E. Sokal, EUTOS и ELTS представлена в таблице II-2. В рекомендациях ELN 2020 г. [16] для использования в практике приведены критерии J. E. Sokal [28] и ELTS [29]. Оценка прогноза общей выживаемости у больных ХМЛ по J. E. Sokal et al. была предложена в 1984 г. и является одной из наиболее первых прогностических систем при ХМЛ. Следует отметить, что для её разработки использовались данные выживаемости больных ХМЛ при проведении сдерживающей терапии цитостатиками, в основном, бусульфаном. Несмотря на это, данная шкала до сих пор позволяет прогнозировать выживаемость больных уже при проведении таргетной терапии и используется во всех клинических исследованиях и входит в международные рекомендации.
Шкала ELTS, напротив, является одной из наиболее новых и разработана по результатам международного многоцентрового исследования, аккумулировавшего результаты лечения больных ХМЛ с использованием таргетной терапии не только в рамках клинических исследований, но и обычной практики. В данной шкале, как оказалось, возраст имеет меньшее значение для прогноза общей выживаемости, чем в шкале Sokal. Таким образом, данная шкала является наиболее релевантной существующей в настоящее время клинической практике. Шкала EUTOS также разработана на основе анализа опыта лечения больных ХМЛ таргетной терапией и позволяет прогнозировать достижение полного цитогенетического ответа на 18 месяцев терапии. Мы рекомендуем её использование в клинической практике благодаря простоте определения прогностической группы с использованием только арифметической суммы произведений размеров селезенки и процента базофилов на соответствующие коэффициенты, что может легко быть выполнено «в уме» непосредственно во время приема пациента.
Таблица II-2. Определение групп риска ХМЛ по J. E. Sokal [28], EUTOS [30] и ELTS [29]
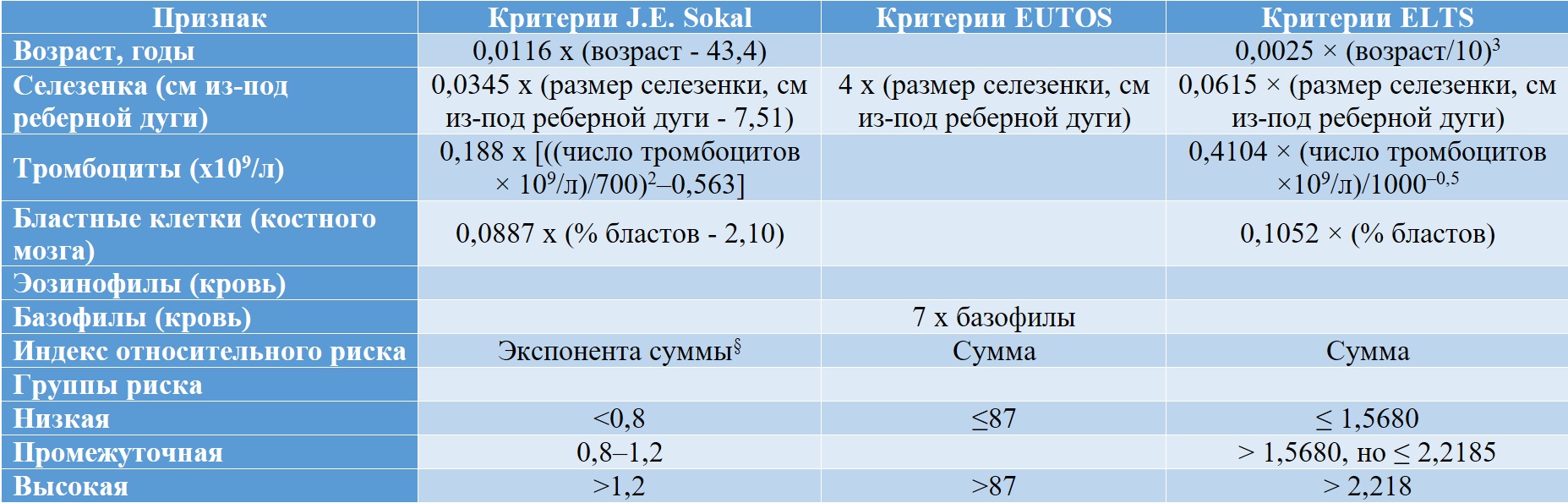
* Автоматический подсчет доступен на сайтах: http://bloodref.com/myeloid/cml/sokal-hasford и http://www.leukemia-net.org/content/leukemias/cml/elts_score/index_eng.html
§ 2,72 в степени (0,0116 x (возраст – 43,4) + 0,0345 x (размер селезенки, см из-под реберной дуги – 7,51) + 0,188 x [((число тромбоцитов × 109/л)/700)2–0,563] + 0,0887 x (% бластов – 2,10))
0,0025 × (возраст / 10)3 + 0,0615 × (размер селезенки, см из-под реберной дуги) + 0,1052 × (% бластов) + 0,4104 × (число тромбоцитов × 109/л)/1000–0,5
Рекомендации ELN 2020 выделяют дополнительные факторы риска прогрессирования ХМЛ в виде фиброза в биоптате костного мозга и дополнительных хромосомных аномалий (ДХА) высокого риска в Ph+ клетках. Такими ДХА считаются, в частности: +8, вторая Ph-хромосома (+Ph), i (17q), +19, – 7 / 7q-, 11q23, 3q26.2, комплексные кариотипы.
Наличие ДХА высокого риска предсказывает более слабый ответ на ИТК и более высокий риск прогрессирования.
В настоящее время ELN 2020 рекомендует классифицировать ДХА и лечить пациентов с ДХА высокого риска как пациентов высокого риска, то есть использовать ИТК2 в первой линии терапии.
По данным Регистра больных ХМЛ в РФ у 6560 (93,8 %) больных заболевание диагностировано в хронической фазе (ХФ), у 380 (5,5 %) – в ФА, у 47 (0,7 %) – в БК. Распределение по прогностической шкале Sokal у 6560 больных в ХФ составило 49:30:21 % для групп низкого, промежуточного и высокого риска соответственно. Наибольшая доля больных с высоким риском по Sokal (до 30 %) наблюдалась среди пациентов старше 60 лет.
Результаты нашего собственного опыта обследования 307 больных ХМЛ показали при первичном обследовании следующее распределение по фазам: ХФ – 282 (92 %) пациентов, ФА – 22 (7 %) больных, БК – 3 (1 %) пациентов. Характеристики пациентов представлены в табл. II-3.
Таблица II-3. Клинические показатели пациентов с ХМЛ на момент первичной диагностики

Распределение по группам риска при диагностике было следующим:
• Sokal низкая 68 % / промежуточная 6 % / высокая 26 %;
• EUTOS низкая 85 % / высокая 15 %;
• ELTS низкая 57 % / промежуточная 30 % / высокая 13 % [31].
Лечение. Цель современной терапии ХМЛ – максимальное подавление Ph-положительного опухолевого клона с восстановлением нормального гемопоэза, что предотвращает прогрессирование заболевания и приводит к продолжительности жизни больных, сравнимой с общей популяцией. Достижение полного цитогенетического ответа (ПЦО) и большого молекулярного ответа (БМО) – это ранние благоприятные прогностические признаки длительной выживаемости без прогрессирования при условии постоянной терапии [19, 30, 32–34]. Новой целью, декларируемой в рекомендациях ELN 2020, является достижение глубокого молекулярного ответа (ГМО), что позволяет, при соблюдении других критериев безопасности, переводить пациентов в так называемую ремиссию без лечения (РБЛ), когда отменяется прием препаратов – ингибиторов тирозинкиназ и пациент остается только под частым молекулярным мониторингом минимальной остаточной болезни. При достижении порогового значения, обычно МО4,0 или БМО, таргетная терапия возобновляется.
При нахождении пациента в РБЛ фактически можно констатировать достижение ремиссии злокачественного заболевания кроветворной ткани, когда нет необходимости приема препаратов, а все медицинские вмешательства по поводу «рака крови» сводятся к контрольному лабораторному обследованию.
После цитогенетического и/или молекулярно-генетического подтверждения диагноза ХМЛ должна быть начата терапия ингибиторами тирозинкиназ (ИТК). Прием ИТК можно начинать при любом уровне лейкоцитов и тромбоцитов. Терапия ИТК в связи с ее таргетным механизмом действия не сопровождается развитием синдрома лизиса опухоли [19, 33].
Характеристика и принципы выбора ИТКЛечение ХМЛ препаратами ИТК коренным образом изменило прогноз этого ранее фатального заболевания, улучшив общую выживаемость в несколько раз и сделав возможной перспективу максимально полного подавления остаточного лейкозного клона. В настоящее время ИТК являются основным средством терапии ХМЛ и имеют доказанное преимущество перед другими методами лечения. Механизм действия ИТК обусловлен блокадой АТФ-связывающего кармана молекулы BCR::ABL, что лишает белок BCR::ABL тирозинкиназной активности, дающей опухолевым клеткам пролиферативное преимущество. При постоянном воздействии ИТК происходит редукция опухолевого клона и восстановление нормального гемопоэза.