
Каталитический риформинг бензинов. Теория и практика
В табл. 2 представлены термодинамические данные для расчета реакции изомеризации н-гексана в 2-метилпентан.
Реакция имеет при 800 К небольшое отрицательное значение ΔгG, что обусловливает величину Kр = 0,8, характерную для реакций изомеризации парафинов.
Ниже представлен расчет равновесного состава смеси с учетом образования всех возможных изомеров н-гексана (рис. 10).

Рис. 10. Схема реакций с образования изомерных гексанов:
2-МП – 2-метилпентан; 3-МП – 3-метилпентан;
2,2-ДМБ – 2,2-диметилбутан; 2,3-ДМБ – 2,3-диметилбутан
В соответствии со схемой реакций в равновесной смеси находится пять компонентов, при этом справедливы следующие соотношения для парциальных давлений компонентов
Рн-г = Р2МП/Kр 2МП = Р3МП/ Kр 3МП = Р2,2ДМБ/ Kр 2,2ДМБ =
= Р2,3ДМБ/ Kр 2,3 ДМБ;
Рнг + Р2МП + Р3МП + Р2,2 ДМБ + Р2,3 ДМБ = Р или
Рнг (1 + Kр 2МП + Kр 3МП + Kр 2,2ДМБ + Kр 2,3ДМБ) = 1.
Отсюда мольные доли компонентов:
Yнг = 1/1 + Kр 2МП + Kр 3МП + Kр 2,2ДМБ + Kр 2,3ДМБ;
Y2МП = Kр 2МП/1 + Kр 2МП + Kр 3МП + Kр 2,2ДМБ + Kр 2,3ДМБ;
Y3МП = Kр 3МП/1 + Kр 2МП + Kр 3МП + Kр 2,2ДМБ + Kр 2,3ДМБ;
Y2,2ДМБ = Kр 2,2ДМБ/1 + Kр 2МП + Kр 3МП + Kр 2,2ДМБ + Kр 2,3ДМБ;
Y2,3ДМБ = Kр 2,3ДМБ /1 + Kр 2МП + Kр 3МП + Kр 2,2ДМБ + Kр 2,3ДМБ.
Результаты расчета равновесных составов для двух температур – 800 К и 700 К, являющихся границами рабочих температур платформинга, – представлены в табл. 3.
Таблица 3
Состав равновесных смесей для двух температур
Компонент
700 К
800 К
н-гексан
2МП
3МП
2,2ДМБ
2,3ДМБ
0,2371
0,3351
0,1809
0,1533
0,0936
0,2692
0,3369
0,1869
0,1215
0,0855
П р и м е ч а н и е : данные в таблице указаны в мольных долях.
Как следует из полученных данных, введение в реакционную схему дополнительных компонентов снижает мольную долю н-гексана при сохранении прочих параметров, что демонстрирует очень сильную зависимость равновесного состава от реакционной схемы. Этот эффект имеет место даже при включении в реакционную схему компонента, образование которого в отдельности термодинамически неблагоприятно, так как и для такого рода реакций Kр > 0, что увеличивает знаменатель в формуле расчета для мольной доли сырьевого компонента.
Интересно отметить, что с увеличением молекулярной массы парафинового углеводорода количество возможных изомеров возрастает, и в связи с этим должно происходить уменьшение равновесной концентрации нормального углеводорода, то есть термодинамическая глубина конверсии парафинов должна увеличиваться с ростом молекулярной массы.
Формально это вытекает из формулы для определения мольной доли компонента, но имеется и более глубокий физический смысл, который состоит в том, что возрастание числа компонентов означает увеличение количества перестановок в химической системе, то есть рост энтропии химической системы в больцмановской интерпретации энтропии. Так, при изомеризации н-гептана (9 компонентов реакционной смеси) равновесное содержание н-гептана при 700 К составляет 0,19 вместо 0,24, полученного для н-гексана (5 компонентов).
Увеличение температуры изомеризации приводит к снижению констант равновесия и равновесной степени превращения в изомеры.
При повышении температуры происходит изменение соотношения между моно-, ди- и триметилзамещенными изомерами с сокращением доли более разветвленных изомеров.
Ниже, на рис. 11, представлено изменение состава реакционной смеси в зависимости от температуры для гептанов.

Рис. 11. Состав смеси для реакции изомеризации
для нормального гептана [50]
В целом вклад реакций изомеризации в увеличение октановых чисел катализата платформинга ограничен из-за высоких температур процесса и является второстепенным фактором, в отличие от промышленных процессов изомеризации н-пентана и гексанов, проводимых при существенно более низких температурах.
Глава 6. Кинетика реакций платформинга
Уравнение Аррениуса.
Быстрые и медленные реакции.
Почему происходит увеличение скорости реакции циклизации при переходе от н-гексана к н-гептану.
Кинетический и термодинамический контроль реакций риформинга.
Структурно-чувствительные и структурно-нечувствительные реакции
Термодинамика устанавливает принципиальные ограничения на направление и максимальную глубину химического превращения. Равновесная степень химического превращения является предельно возможной величиной, которая может быть получена при условии достижения химического равновесия при данных давлении и температуре.
Реально достижимая степень превращения может быть ниже, если достижение химического равновесия невозможно из-за существования кинетических барьеров в виде высоких энергий активации реакции.
Зависимость константы скорости реакции от температуры описывается уравнением Аррениуса, где экспоненциальный множитель представляет собой долю молекул, обладающих кинетической энергией не менее Еа при данной температуре Т:
где Kо – предэкспоненциальный множитель; Еа – энергия активации; R – универсальная газовая постоянная; Т – температура, К.
Значения энергии активации для ряда реакций платформинга, кДж/моль, представлены ниже:
– изомеризация парафиновых и нафтеновых углеводородов – 105,
– дегидрирование парафиновых и нафтеновых углеводородов – 84,
– дегидроциклизация парафинов – 145,
– крекинг – 185,
– коксование – 145 [50].
На рис. 12 приведены значения относительных скоростей реакций платформинга при температуре 500 С для различных парциальных давлений водорода.
Базовым уровнем является скорость дегидрирования, ее значение принято за 100 %.
Крекинг здесь представлен как сумма реакций гидрогенолиза и гидрокрекинга.
Для реакции образования кокса скорость при парциальном давлении водорода 10 бар принята за единицу.
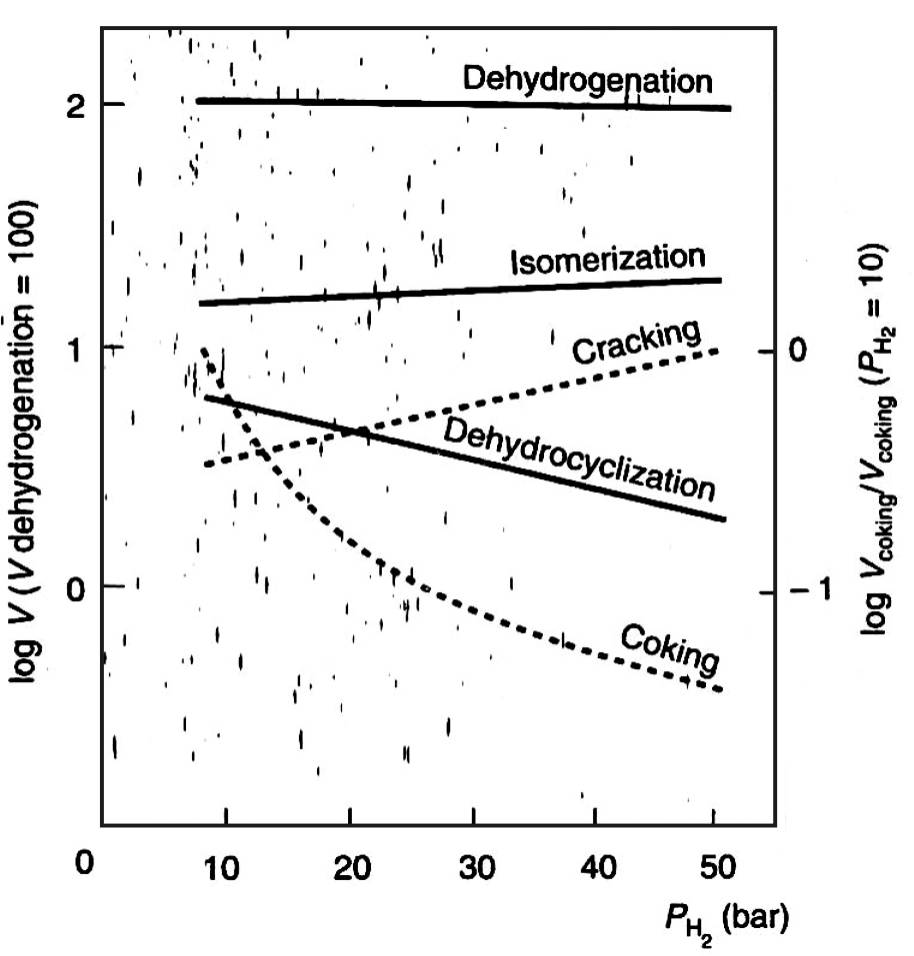
Рис. 12. Скорость реакций платформинга
Дегидрирование нафтенов является самой быстрой реакцией платформинга, ее скорость в 7–8 раз превышает таковую для реакции изомеризации парафиновых и нафтеновых углеводородов и примерно в 30 раз скорость реакций крекинга и дегидроциклизации.
Реакция образования кокса является самой медленной реакцией платформинга.
Для реакций дегидроциклизации и крекинга константы скорости зависят также от длины углеродной цепи и увеличиваются при ее росте.
Особенно резкое увеличение констант скорости наблюдается для реакции дегидроциклизации при переходе от н-гексана к н-гептану, что объясняется статистическим фактором, а именно увеличением количества вариантов замыкания цепи.
Дегидроциклизация н-гексана в условиях бифункционального катализа протекает по схеме:
Лимитирующей стадией этих превращений является циклизация олефина с образованием 5-членного кольца.
Природа высокого энергетического барьера этой реакции может быть обусловлена циклической структурой активированного комплекса, являющегося переходным состоянием химической системы на ее пути от реагентов к продуктам реакции.
В соответствии с теорией активированного комплекса константа скорости реакции
где ΔH# и ΔS# – это изменение энтальпии и энтропии системы при образовании активированного комплекса.
Очевидно, что при образовании циклического комплекса энтропия системы уменьшается. Оценка изменения энтропии может быть сделана по изменению энтропии реакции циклизации.
Результаты представлены ниже в сравнении с изменением энтропии реакции изомеризации н-гексена-1 в 2-метилпентен-1 (табл. 4).
Таблица 4
Изменение термодинамических параметров при 800 К
Параметр
Изомеризация
С5-циклизация
С6-циклизация
ΔrG
ΔrH
ΔrS
–9500
+9400
+0,2
–15 600
–60 300
–56,0
–8300
–84 200
–94,8
Из данных табл. 4 следует, что при циклизации происходит значительное уменьшение энтропии. Применяя эти цифры для активированного комплекса, найдем отношение констант скорости реакции циклизации и изомеризации – 0,0012. Расчет отношения констант скоростей по энергиям активации дает такие же значение – 0,0012. Совпадение скорее случайное, но даже такой грубый расчет показывает, что вклад энтропии образования активированного комплекса может быть определяющим фактором низкой скорости С5-циклизации.
Увеличение скорости циклизации при переходе от н-гексана к н-гептану приводит к существенному увеличению селективности ароматизации алкана.
В табл. 4 представлено также изменение энтропии реакции при С6-циклизации 2-метилпентена-1: в этой реакции происходит еще более значительное уменьшение энтропии.
Если применить аналогичный подход для оценки отношения констант скорости двух альтернативных маршрутов циклизации, получим величину отношения С6/С5, равную 0,01. Это коррелирует с кинетическими данными, в соответствии с которыми С5-циклизация является главным маршрутом дегидроциклизации парафиновых углеводородов риформинга на бифункциональном катализаторе.
Механизм циклизации достоверно не установлен.
В соответствии с гипотезой Гейтса [2], циклизация протекает по согласованному механизму с участием кислотного бренстедовского и основного льюисовского центров:
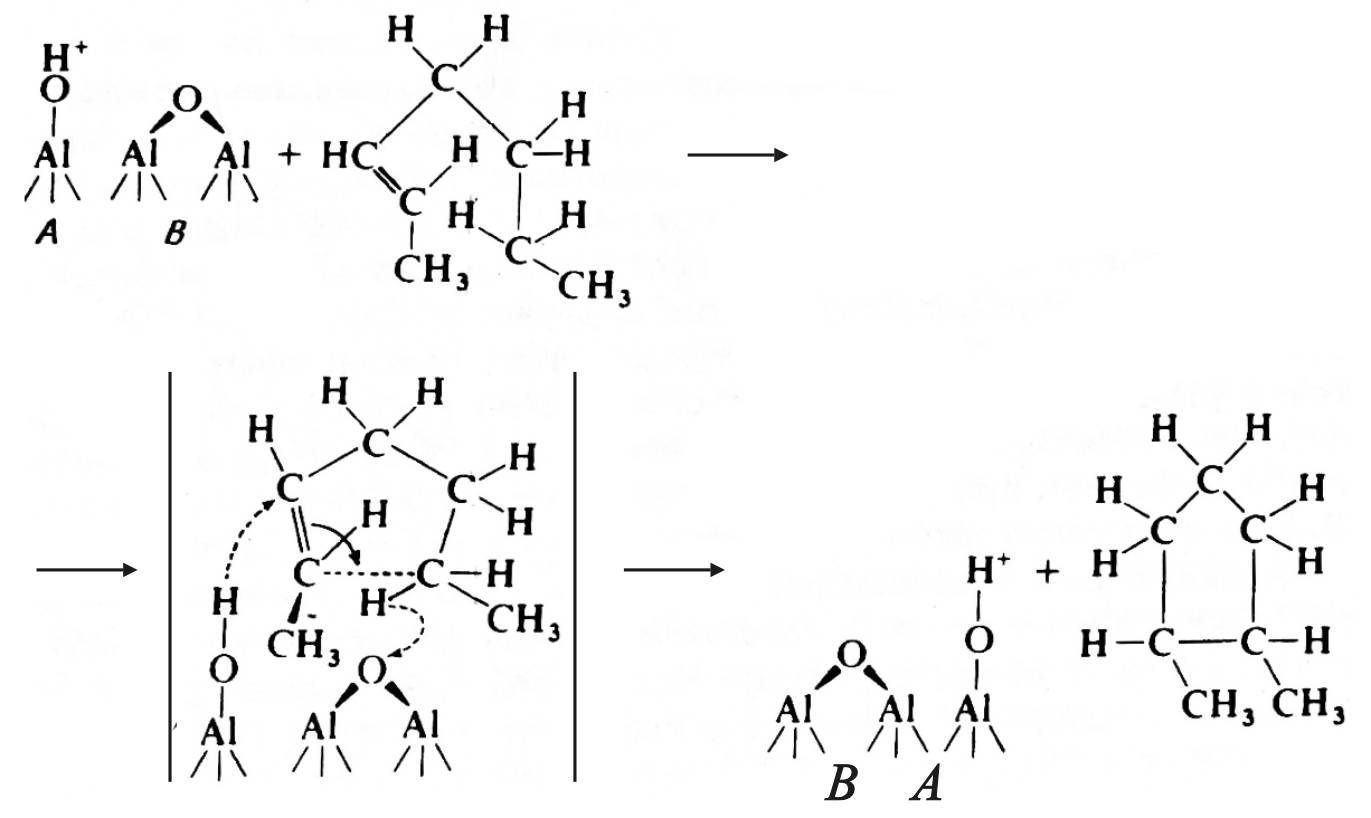
,
где А – кислотный бренстедовский центр; В – льюисовский основный центр.
Альтернативная гипотеза предполагает участие только льюисовских центров и поддерживается рядом экспериментальных фактов: отсутствие эффекта ингибирования азотом реакции циклизации, но ингибирование реакции расширения цикла. Известно также, что реакция циклизации может быть затруднена при увеличении влажности, что связывают с превращением льюисовских центров в бренстедовские.
При термодинамическом контроле направление и выход продуктов химического превращения определяются величиной и знаком изменения энергии Гиббса.
В случае кинетического контроля основным продуктом превращения является продукт реакции с меньшей энергией активации. Примером может служить превращение олефинов на кислотных центрах катализатора риформинга.
Из двух возможных химических реакций – изомеризации и гидрокрекинга – основным продуктом превращения является олефин, хотя его образование сопровождается меньшим понижением энергии химической системы. Превращение олефинов на кислотных центрах в условиях кинетического контроля обеспечивает высокую селективность процесса риформинга. Увеличение кислотности катализатора, повышение температуры процесса, увеличение времени контакта будет благоприятствовать протеканию реакции гидрокрекинга и снижению селективности ароматизации сырья.
Примером превращения, протекающего под термодинамическим контролем, является реакция изомеризации алканов, рассматривающаяся как совокупность параллельных реакций, каждая из которых приводит к образованию определенного изомера. Скорости реакций примерно одинаковы, и состав продуктов суммарного превращения определяется стремлением химической системы минимизировать энергию Гиббса.
В зависимости от того, что контролирует превращение, термодинамика или кинетика, зависит эффект, достигаемый от изменения температуры процесса.
Попытка увеличить скорость реакции изомеризации применением более высокой температуры и получить за счет этого более высокий выход изомеров приводит к обратному результату, так как реакция находится в равновесии, а для равновесной экзотермической реакции увеличение температуры уменьшает Kр.
В случае дегидроциклизации парафиновых углеводородов, еще одного примера кинетического контроля, повышение температуры, наоборот, является основным технологическим приемом интенсификации процесса риформинга парафинистого сырья.
В некоторых случаях желательным является снижение температуры и при осуществлении равновесных эндотермических реакций. Изомеризация метилциклопентена в циклогексен – это равновесная эндотермическая реакция, и для нее
повышение температуры увеличивает конверсию. Однако эта реакция сопровождается крекингом нафтенов, реакцией, контролируемой кинетикой, и для нее увеличение температуры приводит к возрастанию скорости реакции, что означает ухудшение селективности реакции изомеризации. В таком случае снижение температуры в головном реакторе платформинга или проведение процесса при еще более низкой температуре в дополнительном реакторе, устанавливаемом после сырьевого теплообменника или конвекционного змеевика комбинированной печи, позволяет минимизировать нежелательные реакции крекинга и может быть эффективным приемом при переработке нафтенового сырья.
Понятие о структурно-чувствительных и структурно-нечувствительных реакциях риформинга введено Бударом и Тэйлором. К структурно-нечувствительным относят реакции, удельная скорость которых остается постоянной при изменении состава поверхности и размера частиц активной фазы, координационного числа поверхностных атомов, природы носителя.
Реакции гидрирования-дегидрирования алканов и циклоалканов и изомеризации алканов С6 и выше принято относить к структурно-нечувствительным, а реакции гидрокрекинга, гидрогенолиза углерод-углеродной связи, изомеризации алканов С4 и меньше, изомеризации метилциклопентана, дегидроциклизации н-гептана, а также реакции образования кокса являются структурно-чувствительными [47].
Как правило, структурно-чувствительные реакции связаны с разрывом С–С-связей, активирование которых на платине не так успешно, как С–Н-связей. Поэтому эти реакции имеют более высокие энергии активации, что объясняет их предрасположенность к протеканию на ансамблях, состоящих из нескольких адсорбционных центров.
Отнесение реакции к тому или иному типу достаточно условно. Так, в [102] было установлено снижение активности в реакции дегидрирования циклогексана при переходе от 0,3%Pt/Al2O3 к биметаллическим катализаторам 0,3%Pt, 0,3%Re/Al2O3 и 0,3%Pt, 0,3%Sn/Al2O3.
Для Pt–Sn-катализатора снижение составило 23 %.
Учитывая, что содержание платины в катализаторах не менялось, этот результат можно объяснить разбавлением поверхностных атомов платины вторым металлом и уменьшением размера каталитического ансамбля, то есть структурной чувствительностью реакции, обусловленной проведением тестов на активность при сравнительно низкой температуре (400 С), недостаточной для активирования молекулы на одиночном адсорбционном центре.
Глава 7. Режимы протекания
реакций риформинга
Элементарные стадии химического превращения на пористом катализаторе.
Кинетический и внутридиффузионный режимы химических реакций.
Диффузионный модуль Тиле и коэффициент эффективности катализатора.
Режимы протекания реакций риформинга
В гетерогенном катализе большинство промышленных катализаторов являются пористыми материалами, и реакция протекает на внутренней поверхности катализатора.
Химическое превращение является результатом сочетания процессов массопереноса, адсорбции – десорбции и собственно химической реакции.
Для простой реакции
А1 = А2
можно выделить семь основных стадий, представленных на рис. 13 [58].
Массоперенос внутри гранул обеспечивается за счет кнудсеновской и молекулярной диффузии и описывается первым законом Фика
J = –DdC/dx,
где J – удельный поток, моль/м2·с; D – коэффициент диффузии компонента; dC/dx – градиент концентрации вдоль поры.
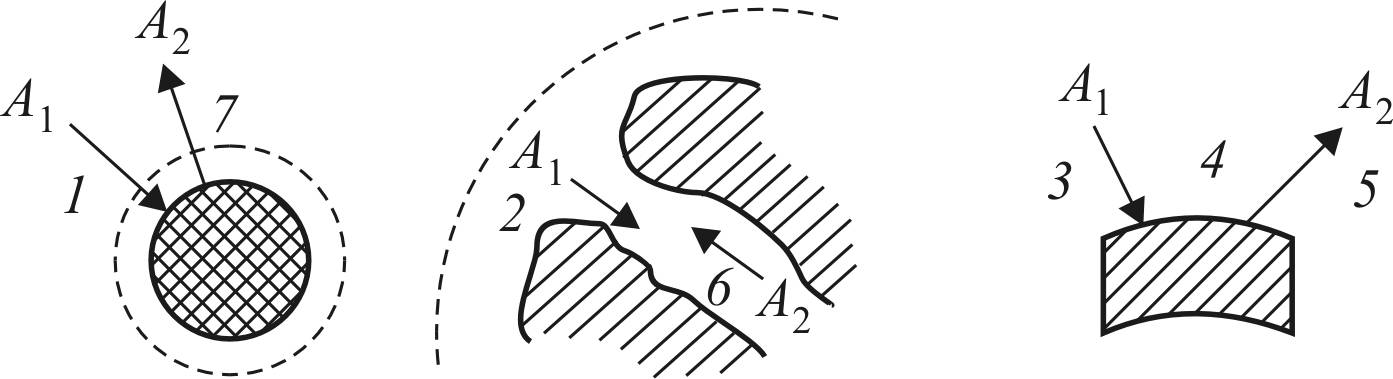
Рис. 13. Основные стадии химического превращения
на гетерогенном катализаторе: 1 – диффузия А1 через пограничный слой, прилегающий к грануле катализатора; 2 – диффузия А1
внутри поры; 3 – адсорбция А1 на активных центрах катализатора;
4 – химическая реакция А1 = А2; 5 – десорбция А2; 6 – диффузия А2
к устью поры; 7 – диффузия А2 через пограничный слой
в ядро потока
Соотношение между молекулярной и кнудсеновской диффузией зависит от отношения средней длины свободного пробега молекул λ и диаметра поры dп. При ограничении длины свободного пробега и λ < dп преобладает массоперенос путем молекулярной диффузии. При λ > dп главным механизмом массопереноса становится кнудсеновская диффузия.
В режиме кнудсеновской диффузии средняя длина свободного пробега сравнима с размером поры, в результате молекулы чаще сталкиваются со стенкой, чем между собой. При контакте со стенкой молекула адсорбируется и какое-то время удерживается на поверхности, покидая ее в произвольном направлении, в том числе навстречу потоку.
Оба явления, адсорбция и диффузное отражение, приводят к снижению коэффициента кнудсеновской диффузии по сравнению с молекулярной [16]:
dк = 9700r (T/M) 1/2,
где dк – коэффициент диффузии, см2/с; r – радиус поры, см;
Т – температура, К; М – молекулярная масса.
Средняя длина свободного пробега может быть определена по формуле
λ = kT/σP,
где k – постоянная Больцмана; Т – температура; Р – давление; σ – эффективное сечение молекулы.
Используя значение σ для молекулы бензола, равное 0,88 нм2, найдем длину свободного пробега при 800 К и давлении 10 бар:
λ = 12,5 нм.
Для большинства пор, обеспечивающих активность катализаторов риформинга, λ не превышает найденного значения, что позволяет сделать вывод о преобладающей роли кнудсеновской диффузии.
Массоперенос с помощью молекулярной диффузии осуществляется в порах большого диаметра и в ламинарном слое газа, окружающего гранулы катализатора. В зависимости от соотношения скоростей массопереноса и химической реакции реализуются различные режимы протекания химического превращения. В общем случае это кинетический режим, внутридиффузионный режим и внешнедиффузионный режим. В условиях платформинга реализуются первые два режима.
В кинетическом режиме скорость превращения определяется скоростью химической реакции (стадии 3–5) (см. рис. 13).
В этом режиме концентрации и температуры внутри гранулы катализатора остаются постоянными, реакция происходит на всей внутренней поверхности гранулы, температурная зависимость превращения определяется энергией активации химической реакции.
При увеличении скорости химической реакции или снижении скорости диффузии кинетический режим переходит во внутридиффузионный, в котором скорость превращения контролируется как химической реакцией, так и диффузией в порах.
При протекании реакции во внутридиффузионном режиме температурная зависимость превращения определяется эффективной энергией активации, представляющей среднее арифметическое действительной энергии активации химической реакции и энергии активации диффузии. В первом приближении, учитывая небольшое значение энергии активации диффузии, эффективная энергия активации примерно равна половине энергии активации химической реакции.
Диффузионный режим характеризуется наличием градиентов температуры и концентраций по сечению гранулы катализатора и по длине поры.
Причиной диффузионных затруднений могут быть повышение температуры каталитического процесса или уменьшение размера пор, или увеличение размера гранулы катализатора.
В зависимости от причины, вызвавшей переход в диффузионный режим, возникают различные последствия.
В первом случае сохраняется зависимость скорости химического превращения от температуры, и более высокой температуре соответствует большая скорость превращения.
Во втором случае при постоянной температуре реакции диффузионные затруднения приводят к уменьшению скорости химического превращения.
Оценка вклада внутридиффузионных эффектов производится при помощи коэффициента эффективности η и диффузионного модуля Тиле φ.
Коэффициент эффективности – отношение фактической скорости химического превращения к скорости, которая могла быть, если бы температура и концентрация реактанта на внутренней поверхности равнялась бы температуре и концентрации на внешней поверхности.
Значение η для изотермической реакции, протекающей во внутридиффузионном режиме, находится в интервале 1 > η > 0.
При η = 1 существует кинетический режим, при η = 0 внутридиффузионный режим переходит в режим, когда химическое превращение не зависит от размера и поровой структуры частицы и лимитируется внешней диффузией.
Диффузионный модуль Тиле для химической реакции первого порядка находится по формуле
φ = Vp/Sp(K/De)1/2,
где Vp и Sp – объем и внешняя поверхность частицы катализатора; K – константа скорости реакции, отнесенная к объему частицы; De – коэффициент эффективной диффузии.
Зависимость коэффициента эффективности от модуля Тиле для изотермической реакции представлена на рис. 14 [58].

Рис. 14. Зависимость коэффициента
эффективности от модуля Тиле
Основные реакции платформинга являются эндотермическими.
Эндотермичность приводит к снижению температуры в грануле катализатора и уменьшению скорости реакции по сравнению с таковой на поверхности гранулы. Это приводит к дополнительному снижению коэффициента эффективности, что демонстрируется на рис. 15.
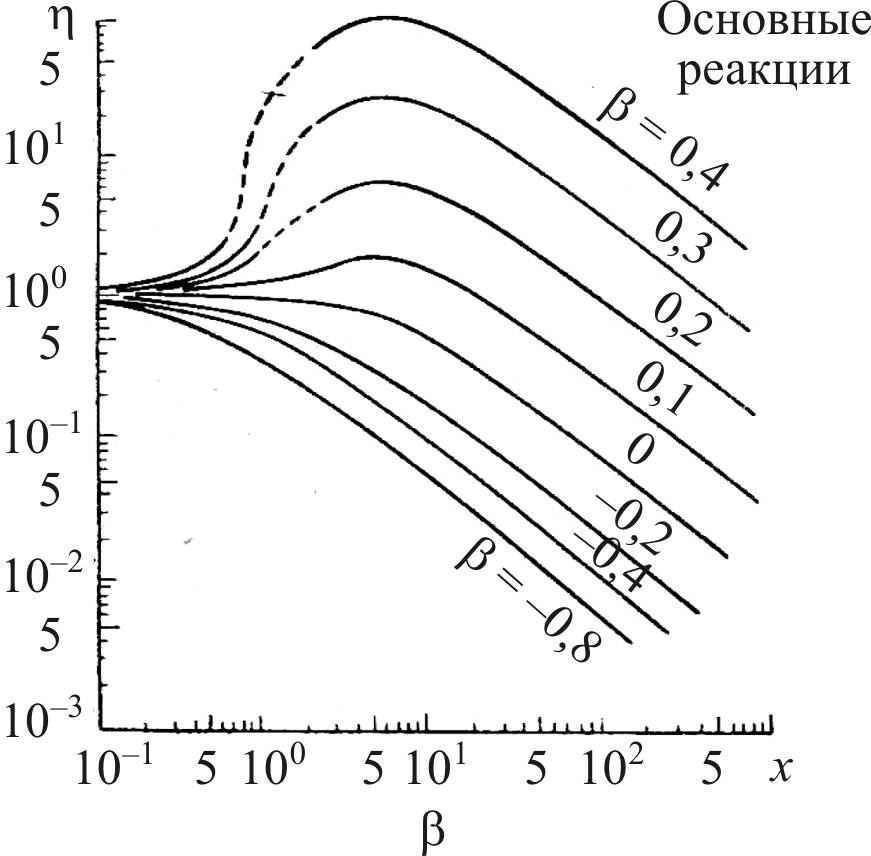
Рис. 15. Изменение коэффициента эффективности в зависимость от β:
β = (–ΔН) DeCs/λTs; ΔT = βTs, где ΔН – энтальпия химической
реакции, De – коэффициент диффузии, Сs – концентрация реагента на поверхности гранулы, λ – коэффициент теплопроводности материала гранулы, Тs – температура на поверхности гранулы,
ΔT – градиент температуры по сечению гранулы, β < 0 для эндотермических реакций и β > 0 для экзотермических реакций
С учетом небольшого размера молекул, участвующих в реакциях риформинга, можно было ожидать, что диффузионные затруднения будут отсутствовать, однако использование катализаторов с высокой удельной поверхностью и, следовательно, с узкими порами, с одной стороны, и высокие скорости реакций, связанные с применением платины и повышенных температур, с другой стороны, объективно создают условия для протекания реакций риформинга во внутридиффузионном режиме.
По данным [17], дегидрирование циклогексана уже при температуре 430 С на платиновом катализаторе с диаметром гранулы 3 мм (размер гранул монометаллических катализаторов риформинга первого поколения) протекает в условиях сильного диффузионного контроля: коэффициент эффективности η составил 0,38. Это означает, что более 60 % удельной поверхности катализатора используется неэффективно из-за сильного падения концентрации циклогексана в центральной части гранулы.
Снижение размера гранулы с 3,0 до 1, 6 мм, применяемых в современных катализаторах, позволяет уменьшить модуль Тиле в 1,9 раза и за счет этого увеличить эффективность использования внутренней поверхности катализатора.
В связи с тем что зависимость константы скорости химической реакции от температуры является экспоненциальной, а температурная зависимость коэффициентов диффузии представляет степенные функции (Т3/2 для молекулярной диффузии и Т1/2 для кнудсеновской диффузии), при рабочих температурах платформинга коэффициент эффективности будет еще ниже.
Оценка c учетом изменения размера гранулы и температуры дает значение η для реакции дегидрирования циклогексана в условиях риформинга, равное 0,31.