
Внутренние болезни. Том 2
Лечение больных ЖДА может проводиться в амбулаторных условиях. Терапией выбора являются препараты для перорального применения. В настоящее время их предложено довольно много. Наиболее эффективные: ферроплекс, конферон, феррокаль, орферон, ферроградумет, фенюльс, гемофер пролонгатум, сорбифер дурулес, тардиферон, апо-ферроглюконат, железа фумарат, мальтофер, феррум лек, венофер и др. Они принимаются сразу после приема пищи из расчета 5 мг/кг массы тела в день. Для большинства препаратов непролонгированного действия максимальная доза препарата 6 таблеток в сутки. В то же время для таких пролонгированных препаратов, как ферроградумет и фенюльс, достаточно и одной таблетки в сутки. В качестве усиливающего всасывание железа средства может быть использована аскорбиновая кислота в дозе 200 мг на 1 г железа.
На фоне приема препаратов железа самочувствие больного быстро улучшается, несмотря на то что уровень гемоглобина поднимается довольно медленно.
Этот парадоксальный эффект связан с общим укрепляющим действием железа на организм, в том числе в плане повышения миоглобина, цитохромов и т. д. Лечение больного с доказанной железодефицитной анемией должно проводиться непрерывно в течение 3 – 4 мес. и быть направлено не только на нормализацию гемоглобина, но и устранение всех симптомов сидеропении и создание депо железа. При отсутствии эффекта от пероральных препаратов показаны парентеральные (феррум лек 100 мг внутримышечно, мальтофер, венофер и т. д.). Однако из-за возможной индивидуальной непереносимости вводить их следует осторожно, а начинать лечение с половинной дозы.
Профилактический прием препаратов железа необходим донорам, беременным, кормящим, а также обильно менструирующим женщинам. В последнем случае желателен ежемесячный (2 – 4 дня) прием препаратов железа внутрь.
Анемия с нарушением синтеза порфиринов (сидеробластная)Второй вид гипохромной анемии – так называемая сидеробластная, или сидероахрестическая анемия. Она связана с нарушением синтеза порфиринов и характеризуется наличием в крови гипохромных микроцитарных эритроцитов, а в костном мозге – кольцевидных сидеробластов. Встречается это заболевание довольно редко. В части наблюдений оно имеет отчетливую наследственную природу, в части носит приобретенный характер.
Этиология. Наследование врожденной формы сидеробластной анемии связано с X-хромосомой и поэтому чаще наблюдается у мужчин. Выделяют также специальный «пиридоксинчувствительный» вариант врожденной сидеробластной анемии, при котором может быть достигнута частичная гематологическая ремиссия на фоне высоких доз витамина В6 (2 – 4 мг/кг/сут).
Приобретенная сидеробластная анемия обычно возникает без отчетливой связи с внешними факторами или заболеваниями. У части больных сидеробластную анемию могут провоцировать: свинцовая интоксикация, длительный прием алкоголя, лечение изониазидом и левомицетином. Кроме того, идиопатическая приобретенная сидеробластная анемия может быть предстадией лейкоза, о чем пойдет речь в разделе «Миелодиспластические синдромы».
Патогенез сидеробластной анемии связан с дефектом синтеза гема в митохондриях эритроидных клеток. Речь идет о нарушении функции синтетазы дельта-аминолевулиновой кислоты и ряда других митохондриальных ферментов. Основным предшественником дельта-аминолевулиновой кислоты в клетке являются сукцинил-коэнзим А и глицин. Ее синтез стимулируется эритропоэтином, а катализируется производным витамина B6 – пиридоксаль-5-фосфатом. Образующийся при этом из дельта-аминолевулиновой кислоты протопорфирин в ходе дальнейших превращений соединяется с двухвалентным железом (Fe2+) и формирует гем. В свою очередь, каждая молекула гема на полирибосомах связывается с глобиновой цепью. Комбинация же четырех цепей глобина с гемом и представляет собой наш гемоглобин.
Если представить теперь, что образование дельта-аминолевулиновой кислоты и протопорфиринов в эритроидных клетках будет нарушено, это приведет, с одной стороны, к накоплению в митохондриях двухвалентного железа, с другой – к недостаточному синтезу гемоглобина. В итоге разовьется анемия, при которой будут иметь место и гиперсидеринемия, и резкое увеличение в костном мозге содержания кольцевидных сидеробластов (нормобластов с расположенными вокруг ядра – в митохондриях – гранулами железа), которые могут быть выявлены специальными красителями. В зависимости от характера анемии (врожденная или приобретенная), а также от непосредственной ее причины механизмы повреждения синтеза гема могут различаться. Так, в случае свинцовой интоксикации наряду с нарушением синтеза гема и глобина происходит отчетливое торможение работы фермента пиримидин-5S-нуклеотидазы, что, с одной стороны, приводит к кумуляции в эритроцитах денатурированной РНК, с другой – к появлению в них базофильной пунктации.
Клиническая картина заболевания проявляет себя анемическим синдромом разной степени тяжести. Некоторые больные нуждаются в трансфузиях эритроцитарной массы. Вследствие неэффективного эритропоэза и внутрикостномозгового гемолиза у них может иметь место некоторая желтушность кожи и склер. Кроме того, нередко определяются умеренная спленомегалия и гепатомегалия. Наконец, у больных с врожденным вариантом могут быть зафиксированы такие признаки гемохроматоза, как гиперпигментация кожи, диабет, дисфункция печени и сердца и т. д. В случае со свинцовой интоксикацией на первое место в клинике могут выступать такие ее яркие проявления, как сильные схваткообразные боли в животе, явления полиневрита, изменение со стороны слизистой полости рта (свинцовая кайма на деснах).
Диагноз сидеробластной анемии должен обсуждаться врачами в случае хорошо верифицированной гипохромной анемии без клинических проявлений сидеропении и при наличии в сыворотке крови высокого содержания железа и ферритина, а также низкого содержания ненасыщенного сидерофилина. Подтверждается диагноз исследованием пунктата костного мозга, выявляющего большое количество кольцевидных сидеробластов. При этом может быть заказан цитoгeнeтичecкий анализ клеток костного мозга, который в случае выявления клоновых изменений кариотипа позволит уверенно диагностировать один из трудно определяемых обычными способами подвариантов миелодиспластического синдрома, а именно рефрактерную сидеробластную анемию, и выбрать правильную тактику ее ведения.
Дифференциальный диагноз сидеробластной анемии проводится с другими видами анемий, и прежде всего с гипохромными. В отличие от железодефицитной анемии, при этой патологии в клинике не будут представлены симптомы сидеропении, а в лабораторных анализах уровень железа не только не снижен, а даже повышен. В отличие от талассемии, дефекты костной системы, так же как спленомегалия, для сидеробластной анемии не характерны. В трудных случаях решающим в распознавании сидеробластной анемии от других анемий может быть высокое содержание в костном мозге кольцевидных сидеробластов, а в ряде случаев также положительный эффект терапии большими дозами витамина В6.
Лечение сидеробластной анемии вызывает у врача много проблем. При наличии выраженной анемии показана заместительная терапия эритроцитарной массой, дополняемая введением десферала для профилактики гемосидероза. При исключении миелодиспластического синдрома оправданна также длительная (до 2 мес.) терапия большими дозами (2 – 4 мг/кг/сут) витамина В6. К сожалению, утешительного эффекта данной терапии удается достичь только у единичных больных с врожденной пиридоксин-чувствительной сидеробластной анемией.
Если такая терапия неэффективна, могут быть предложены для лечения большие дозы андрогенов.
Для профилактики развития гемохроматоза и его осложнений при высоких показателях ферритина можно использовать хелаторы железа (десферал или эксиджад). Описаны также единичные случаи аллогенной трансплантации гемопоэтических стволовых клеток для лечения врожденных форм сидеробластной анемии.
Прогноз заболевания остается крайне серьезным. Основными причинами смерти больных являются осложнения со стороны сердечно-сосудистой и эндокринной систем, печени и почек, в том числе спровоцированные гемохроматозом.
ТалассемииДругим видом гипохромных анемий являются талассемии, которые представляют собой сборную группу заболеваний наследственной природы, при которых нарушен синтез одной или более структурно неизмененных цепей глобина. Количество последних у человека достигает четырех. Отсюда возможны четыре вида талассемии, из которых больший клинический интерес представляют áá-талассемия и â-талассемия. Первая больше распространена в Китае, Индии и на Дальнем Востоке, вторая – в Средиземноморье, Малой и Средней Азии.
á-Талассемия. Подавляющее большинство á-талассемий связано с делецией генов á-цепей глобина, находящихся на хромосоме 16, а клинические и гематологические проявления зависят от количества пораженных генов. При вовлечении одного из четырех генов á-цепей глобина (á-/áá) их носительство не сопровождается ни клиническими, ни гематологическими изменениями. При поражении двух из четырех генов á-цепей глобина (á-/á— или – /áá) определяется микроцитоз и гипохромия, хотя выраженной анемии нет. Делеция трех генов áá-цепей глобина (-/á-) сопровождается развитием гипохромной микроцитарной анемии с мишеневидными эритроцитами (см. цв. вкл., рис. 5.2), тельцами Гейнца в них и умеренным гемолизом. Несмотря на такие морфологические изменения эритроцитов, тяжелого анемического синдрома не развивается, что чаще всего позволяет обходиться даже без гемотрансфузий. Полное отсутствие генов á-цепей несовместимо с жизнью.
â-Талассемия. В отличие от двух генов á-цепи глобина человек наследует от родителей только по одному гену â-цепи, которые расположены на хромосоме 11. Отсюда возможны гетерозиготные, гомозиготные и двойные гетерозиготные варианты талассемии с уменьшением в крови нормального, состоящего из двух áá— и двух â-цепей, гемоглобина Нb А и увеличением гемоглобинов Нb F и Нb А2.
Патогенез. Молекулярные механизмы патогенеза â-талассемии сложнее и разнообразнее, чем при á-талассемии. Так, делеции генов при этом виде талассемии не бывает. Вместо этого выявляются точечные мутации, которых на сегодняшний день описано более 100. В результате мутаций при гетерозиготной талассемии возникает дисбаланс синтеза á-иâ-цепей глобина; развивается неэффективный эритропоэз и гемолиз различной степени тяжести. При гомозиготной ââ-талассемии эти процессы выражены значительно сильнее и проявляют себя как костномозговым разрушением эритроидных элементов, так и массивным повреждением эритроцитов в селезенке. Как следствие этого, развиваются экстрамедуллярные очаги компенсаторного кроветворения.
Клиническая картина талассемии разнообразна. Для «большой» гомозиготной â-талассемии характерен выраженный анемический синдром, который выявляется очень рано (в возрасте 3 – 6 мес.). Из-за гемолиза эти дети отстают в развитии, имеют место деформации костей черепа, истончение коркового вещества костей, спонтанные переломы. Характерна окраска кожи, обусловленная бледностью, желтухой и отложением меланина. Резко увеличены размеры селезенки и печени. Выявляется кардиомегалия и признаки сердечной недостаточности. Могут быть клинические проявления гемохроматоза.
Клинические проявления гетерозиготной â-талассемии менее ярки. В большинстве случаев заболевание выявляется случайно. Оно проявляет себя умеренно выраженным анемическим синдромом, реже небольшой спленомегалией. Желтуха встречается редко.
Лабораторная диагностика талассемии зависит от варианта заболевания. У больных гетерозиготной á-талассемией выявляется разной степени выраженности гипохромная микроцитарная анемия. Встречаются отдельные мишеневидные эритроциты, а также клетки с базофильной пунктацией. Содержание ретикулоцитов и сывороточного железа обычно нормальное. Электрофорез гемоглобина выявляет двухкратное увеличение количества гемоглобина А2, которого при áá-талассемии и других гипохромных анемиях нет. У половины больных может быть также повышен фетальный Нb F.
Приблизительно такие же лабораторные признаки находят при малой á-талассемии. Однако электрофорез гемоглобина патологии не выявляет. Для á-талассемии с поражением 3 генов цепей глобина (болезнь Нb Н) характерно наличие в клетках гемоглобина Н на фоне снижения количества гемоглобина А. Для гомозиготной â-талассемии характерны выраженная гипохромная анемия, увеличение содержания ретикулоцитов в крови, нормоцитоз, мишеневидные эритроциты и их базофильная пунктация. При электрофорезе гемоглобина выявляется резкое снижение или отсутствие гемоглобина А при значительном увеличении гемоглобина F. Кроме того, может быть умеренно увеличено количество гемоглобина А2.
Дифференциальный диагноз талассемии проводится с другими гемолитическими и гипохромными анемиями. В отличие от ЖДА, у больных талассемией отсутствуют синдромы сидеропении, не снижено содержание железа и ферритина в сыворотке крови и, наоборот, не наблюдается увеличение уровня ненасыщенного сидерофилина. В отличие от других гемолитических анемий, при талассемиях, за редким исключением, имеют место гипохромные микроцитарные эритроциты. В трудных случаях помогает электрофорез гемоглобинов, выявляющий их аномальные формы.
Лечение. Больные малыми талассемиями в специальной терапии не нуждаются. Лечение больших талассемий включает: трансфузии эритроцитов после индивидуального подбора доноров по системе HLA, прием фолиевой кислоты (достаточно 5 мг/сут), введение десферала внутривенно 1 – 2 г на каждые 500 мл донорской крови и аскорбиновой кислоты (200 мг/сут), спленэктомию, лечение органных поражений, связанных с гемосидерозом. При наличии HLA-совместимых доноров костного мозга молодым больным (до 16 лет) может быть проведена аллогенная трансплантация, после которой безрецидивная выживаемость достигает 80 %.
Прогноз для больных малыми формами талассемии благоприятен, а для гомозиготной â-талассемии серьезен. Ее основными осложнениями, кроме замедленного развития ребенка, являются инфекции, в том числе вирусные гепатитыВиС,игемосидероз внутренних органов, связанный с множественными гемотрансфузиями.
5.2. АНЕМИИ ПРИ ХРОНИЧЕСКИХ ЗАБОЛЕВАНИЯХ
Определение. Анемию, ассоциированную с инфекционными и воспалительными процессами и онкологией, называют анемией хронического заболевания или анемией на фоне хронического заболевания. Поначалу казалось, что они связаны с нарушенным усвоением железа участвующими в воспалении макрофагами. Однако позднее проявился мультифакториальный генез анемии хронического воспаления, при которых помимо нарушения усвоения железа были еще представлены: а) укорочение длительности жизни эритроцитов; б) снижение в крови уровня эритропоэтина.
Патогенез. Создается впечатление, что ведущую роль в патогенезе этих анемий играет гиперпродукция различных цитокинов, прежде всего фактора некроза опухоли, интерлейкинов 1 и 6, а также интерферонов á, â и ã. В основе развития анемического синдрома при анемии хронического заболевания лежит уменьшение длительности жизни эритроцитов и недостаточная компенсаторная реакция эритропоэза, что обусловлено ингибирующим влиянием цитокинов на эритропоэз и чувствительностью эритроидных клеток-предшественников к эритропоэтину. Усвоение же железа эритроидными клетками-предшественниками может быть не изменено или нарушено, что может являться дополнительным фактором развития анемии. Отсюда понятно, что из этой группы должны быть исключены анемии, обусловленные замещением костного мозга опухолевыми клетками, потерей крови, заболеваниями печени, почек, почечной недостаточностью и эндокринопатиями, хотя многие из этих заболеваний по своей сути тоже хронические.
Распространенность анемий на фоне хронического заболевания. Учитывая большую распространенность среди населения хронических заболеваний воспалительного или опухолевого генеза, эти виды анемии встречаются довольно часто. Основными их причинами являются: а) все хронические инфекции; б) хронические неинфекционные воспалительные заболевания, в том числе ревматоидный артрит, ревматическая лихорадка, системная красная волчанка, тяжелая травма, ожоги, васкулиты и др.; в) злокачественные опухоли и гемобластозы; г) алкогольное поражение печени; д) тяжелая сердечная недостаточность; и др.
Клинические проявления у больных анемией и ее тяжесть в подавляющем числе наблюдений зависят от тяжести основной патологии. Чаще всего на ранних стадиях заболевания имеет место легкая, реже – средней тяжести анемия с колебаниями гематокрита от 0,25 до 0,4, что напрямую зависит от колебаний объема циркулирующей крови и объема циркулирующей плазмы. При высоком уровне в крови интерлейкина 6 на анемию хронического заболевания может наслаиваться «анемия разведения».
Лабораторная диагностика. Для анемии хронического заболевания типично нормальное или сниженное содержание ретикулоцитов в крови. Анемия обычно носит нормоцитарный и нормохромный характер, а в 23 – 50 % случаев может быть микроцитарной и гипохромной. Последний симптом встречается чаще, чем микроцитоз эритроцитов, и нередко может быть одним из первых признаков анемии хронического заболевания.
Также для анемии хронического заболевания характерно снижение показателя сывороточного железа в отсутствие увеличения или даже при наличии снижения общей железосвязывающей способности сыворотки. При этом показатель насыщения трансферрина чаще находится на субнормальном уровне. Снижение сывороточного железа отмечается в ранние сроки тяжелого инфекционного процесса и опережает развитие анемии. В свою очередь, это сопровождается умеренным снижением в костном мозге числа сидеробластов (до 5 – 15 %) и увеличением гемосидерина в макрофагах. Уровень ферритина, как правило, повышен, хотя при сопутствующей ЖДА, которая встречается в 20 % наблюдений, может стать относительно низким.
Важным показателем анемии хронического заболевания является увеличение содержания в крови таких острофазовых белков, как фибриноген, церулоплазмин, гаптоглобин, СРБ, С3-компонент комплемента и амилоид А протеин. Часто у больных анемией хронического заболевания имеет место увеличение туморнекротического фактора, интерлейкинов 1 и/или 6 и интерферонов (прежде всего â-иã-интерферонов). В то же время уровни альбумина и трансферрина, как правило, снижены.
Диагноз анемии на фоне хронического воспаления ставится по данным отмеченного выше нарушения обмена железа, сочетающегося с одной или несколькими из перечисленных выше патологий.
Дифференциальный диагноз, прежде всего, проводится с железодефицитными анемиями. В отличие от ЖДА, при анемии хронического заболевания на фоне снижения содержания железа в сыворотке отсутствует снижение запасов железа в ретикулоэндотелиальной системе и сидеропенический синдром. Кроме того, уровень ферритина в сыворотке крови при этих видах анемий, как правило, повышен.
Лечение. Основой терапии анемии, обусловленной хроническими воспалительными заболеваниями, является лечение основной патологии. В том случае, если из-за анемического синдрома тяжесть основного патологического процесса усугубляется, т. е. усиливается гипоксемия, значимо снижается качество жизни, есть все основания для проведения терапии рекомбинантным эритропоэтином. Стандартная схема лечения подразумевает применение эритропоэтина (эпрекс, рекормон и др.) по 30 МЕ 3 раза в неделю. При этом повышение гемоглобина может быть зафиксировано уже через 3 – 4 нед. Если же эффекта от применения эритропоэтина нет или снижение гемоглобина достигает критических цифр (< 50 г/л), то по жизненным показаниям следует применять трансфузии эритроцитарной массы.
5.3. МАКРОЦИТАРНЫЕ (МЕГАЛОБЛАСТНЫЕ) АНЕМИИ
Мегалобластные анемии представляют собой группу заболеваний, для которых характерны связанные с нарушением синтеза ДНК мегалобластное кроветворение и макроцитоз крови. В основе заболеваний лежит нарушение естественных путей синтеза пуринов и пиримидинов и торможение ДНК-полимеризации. Основными причинами развития макроцитарных анемий считаются дефицит фолиевой кислоты и/или витамина В12.
Этиология и патогенез. Роль витамина В12 и фолиевой кислоты показана на схеме 5.1, из которой видно, что недостаток фолиевой кислоты, и в частности 5, 10-метил-тетрагидрофолат-полиглутамата, приводит к торможению превращения дезоксиуридин монофосфата в дезокситимидин монофосфат и в результате – к торможению синтеза ДНК. В то же время витамин В12 прямого участия в синтезе ДНК не принимает. Однако он необходим клетке для превращения поступающего из кишечника 5-метил-тетрагидрофолата (фолиевой кислоты) в активные коэнзимные формы, в том числе упомянутого выше полиглутамата.
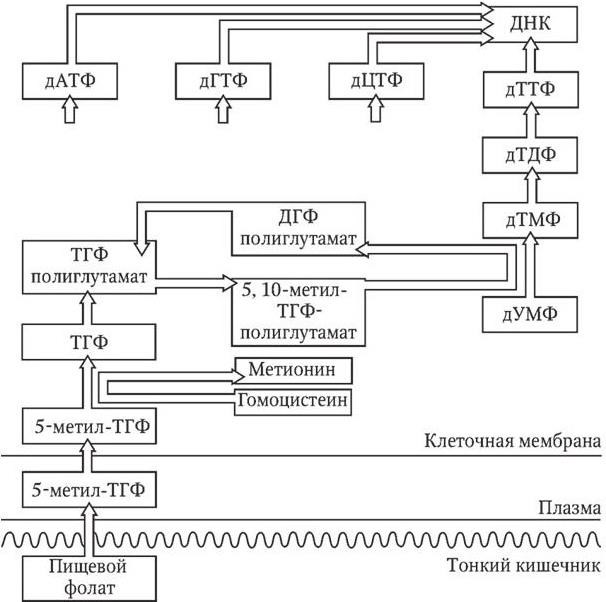
Схема 5.1. Место фолиевой кислоты и витамина В12 в синтезе ДНК, связанное с превращением дезоксиуридинмонофосфата (дУМФ) в дезокситимидинмонофосфат (дТМФ) и далее дезокситимидиндифосфат (дТДФ), дезокситимидинтрифосфат (дТТФ) и тимидин:
ДГФ – дигидрофолат; ТГФ – тетрагидрофолат; дАТФ – дезоксиаденозинтрифосфат; дГТФ – дезоксигуанитидинтрифосфат; дЦТФ – дезоксицитидинтрифосфат
Наиболее частой причиной дефицита фолиевой кислоты является неадекватное поступление ее с пищей, в первую очередь на фоне повышенной потребности. Последняя имеет место у беременных женщин и кормящих матерей, у быстрорастущих детей, особенно в условиях искусственного вскармливания, при гемолизе, некоторых видах злокачественных и воспалительных заболеваний (эксфолиативный дерматит, псориаз, болезнь Крона и т. д.). Другой причиной дефицита фолиевой кислоты может быть нарушение ее всасывания в кишечнике (болезнь Крона, спру) и плохое усвоение. Последнее может иметь место при длительном приеме некоторых противосудорожных (дифенин, барбитураты), противотуберкулезных (циклосерин), противозачаточных и противодиабетических (метформин) препаратов и алкоголя. Наконец, дефицит фолиевой кислоты может возникнуть в результате приема больным прямого ингибитора дигидрофолатредуктазы – метотрексата.
Основной причиной дефицита в организме витамина В12 следует считать нарушение его всасывания в кишечнике из-за недостаточной выработки желудком фактора Кастла. Последнее имеет место у больных атрофическим гастритом, с опухолью желудка, а также может быть следствием ранее перенесенных гастрэктомий и серьезных заболеваний тонкого кишечника. Другой причиной дефицита витамина В12 может быть носительство широкого лентеца, который поглощает этот витамин в большом количестве. Значительно реже дефицит витамина В12 возникает из-за недостатка его в пище, например, у строгих вегетарианцев. Кроме того, нарушение всасывания и усвоения витамина В12, как и фолиевой кислоты, может наблюдаться на фоне приема противотуберкулезных (ПАСК) и противодиабетических (метформин) препаратов, некоторых антибиотиков (неомицин) и алкоголя. Кратковременное снижение активности витамина В12 можно наблюдать в случае применения анестезии закисью азота. Наконец, у новорожденных мегалобластная анемия может возникнуть из-за дефицита в сыворотке транскобаломина II.
Хорошо известно, что основными источниками фолиевой кислоты являются листья растений, а витамина B12 – продукты животного происхождения (мясо, печень, рыба). Вместе с тем его довольно много в сое и некоторых морских водорослях (хиджики, морская капуста). Наконец, он может синтезироваться многими микроорганизмами, в том числе населяющими наш кишечник. Запасов витамина B12 в организме обычно хватает на 5 – 6 лет, а фолиевой кислоты – на несколько месяцев. В желудке витамин В12 высвобождается из белковых комплексов. Далее он связывается с упомянутым выше гликопротеином – внутренним фактором Кастла, который синтезируется париетальными клетками слизистой оболочки желудка. Этот комплекс «витамин/фактор Кастла» соединяется со специфическими рецепторами в дистальных отделах подвздошной кишки, откуда витамин В12 поступает в портальный кровоток в связи с его основным переносчиком – транскобаламином II. При этом внутренний фактор в кровоток не поступает.
Клиническая картина. Мегалобластная В12-дефицитная анемия встречается чаще у женщин, чем у мужчин (1,6: 1,0). Средний возраст больных – старше 60 лет. Поскольку развитие основной причины В12-дефицитной анемии – атрофического гастрита – носит аутоиммунный характер, это заболевание часто сочетается у больных с аутоиммунным тиреоидитом, болезнью Грейвса, иммунным гипопаратиреозом, витилиго, болезнью Адиссона и гипогаммаглобулинемией. Замечено также, что эта анемия может быть уделом лиц, имеющих группу крови II (А), голубые глаза, и обычно сопровождается ранним поседением. У большинства больных В12-дефицитными анемиями (до 90 %) могут быть обнаружены антитела к париетальным клеткам слизистой желудка, у половины – к внутреннему фактору Кастла. С одной стороны, они могут препятствовать связыванию витамина B12 с внутренним фактором, с другой – препятствовать его всасыванию в подвздошной кишке. Следует также заметить, что антитела к внутреннему фактору Кастла для B12-дефицитных анемий являются специфическими, в то время как антитела к париетальным клеткам желудка у части пожилых женщин могут быть обнаружены и в отсутствие анемии.